Chapter: Biotechnology Applying the Genetic Revolution: Proteomics
Peptide Sequencing Using Mass Spectrometry
PEPTIDE
SEQUENCING USING MASS SPECTROMETRY
Determining the peptide
sequence of a short peptide is readily achieved using current mass spectroscopy
techniques (Fig. 9.12). Determining the entire sequence of a protein is too
complex at this point, but may someday be feasible. To determine the sequence,
a pure sample of the protein must be obtained either by cutting a spot from a
two-dimensional gel or by HPLC purification. The protein is then digested into
fragments using a protease such as trypsin, which cuts proteins on the
carboxy-terminal side of arginine and lysine. Cutting a protein into peptides
helps reduce undesirable characteristics of the entire protein.
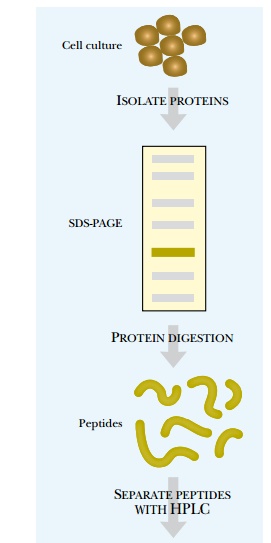
For example, membrane
proteins are hydrophobic and stick together, and digesting these into peptide
fragments destroys this characteristic. Solubility issues can also often be
resolved by digesting a protein into peptides. Determining the sequence of
these peptides will yield the sequence of the original protein.
The most common method of
determining peptide sequences begins by separating the peptides using an HPLC
column directly attached to the mass spectrometer. The column is a microscale
capillary in order to keep the sample volume as small as possible. The mobile
phase is an organic solvent that elutes the peptides in order of their
hydrophobicity.
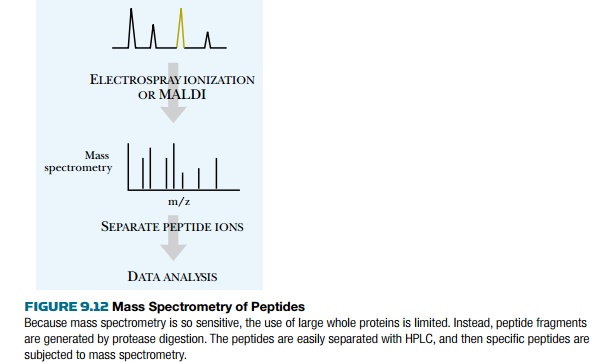
From the HPLC capillary tube,
the samples enter the mass spectrometer chamber. Each peptide is ionized into
multiple fragments (Fig. 9.13). For peptides, common ions include a doubly
protonated form (M + 2H)2+, where M is
the mass of the peptide and H+ is the mass of a proton. The ion
peaks are plotted versus the mass to charge ratio or m/z. For the doubly
protonated peptide ion, this would be the mass of the ion divided by 2. For
example, if the original peptide was 1232.55 daltons, the double protonated ion
would have a mass of 1232.55 daltons + (2 × 1.0073) for each added hydrogen. The peak
would appear at 617.2828. (Note: The mass to charge ratio is where the peak is
plotted, that is, the mass for this ion is 1234.5646 and the charge is +2. The
peak appears at m/z or 1234.5646/2.) When the mass spectrometer separates
peptide ions, the first step is to determine the charge state of the ion.
Usually a cluster of peaks occurs for each peptide ion. If peaks are 1 dalton
apart, the charge state of the peptide is 1. If the peaks are 0.5 dalton apart,
the charge state is 2.
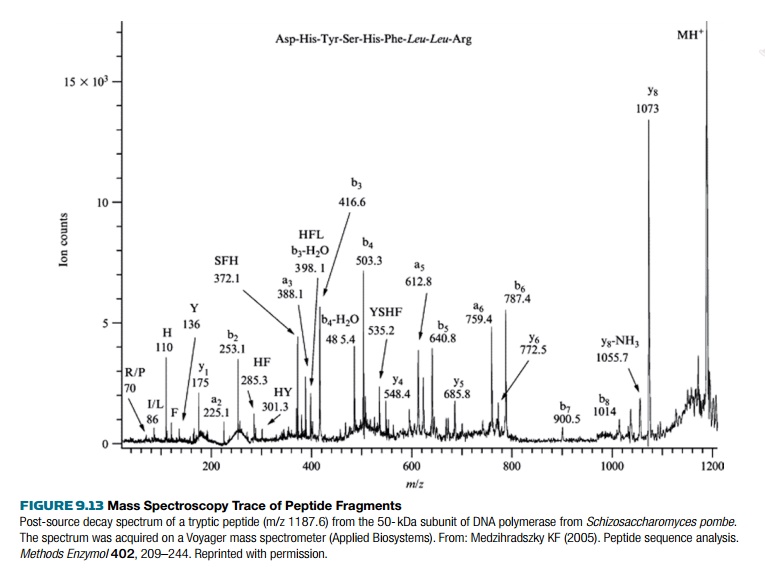
To determine the peptide sequence, two rounds of mass spectroscopy are used. This is called tandem mass spectroscopy because one ion is produced in the first round of mass spectroscopy, then that ion is fragmented by collision with a gas such as hydrogen, argon, or helium. As before, the ion fragments are separated based on their mass to charge ratio. Each peak usually varies by one amino acid, and the size difference between the peaks determines the amino acid sequence. Sometimes the spectrum obtained for a peptide ion is ambiguous, so databases of peptide ion spectra are used for comparison.
Related Topics