Chapter: Biotechnology Applying the Genetic Revolution: Proteomics
Gel Electrophoresis of Proteins
GEL
ELECTROPHORESIS OF PROTEINS
Studying proteins requires
the ability to isolate and identify the proteins in a particular sample. The
first step is to separate them. Much as electrophoresis on agarose gels is used
to separate DNA by size, so polyacrylamide
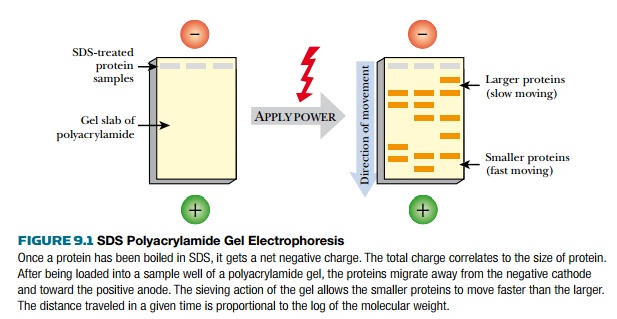
gel electrophoresis (PAGE) is used to separate proteins by size (Fig. 9.1).
Polyacrylamide has smaller pores than agarose and is thus suitable for
proteins, because these are generally smaller than DNA molecules. When an
electric field is applied to a sample of proteins, those with a smaller size
are able to circumnavigate the pores of the acrylamide more easily and will
migrate away from the negative pole faster than larger proteins.
Unlike DNA, most proteins do
not have a net negative charge; indeed, some have a positive charge. Therefore,
protein samples are treated by boiling with sodium dodecyl sulfate (SDS).
This unfolds the polypeptide chains and the entire strand of amino acids is
coated with negatively charged SDS.
The amount of SDS, and therefore the amount of charge, correlates with the
length (i.e., molecular weight) of the protein. As in DNA electrophoresis,
the protein sample is loaded
into a well, and an electric current is applied so that the proteins migrate
through the acrylamide gel. Finally, the separated proteins are visualized
using either Coomassie Blue, a dark
blue dye, or the more sensitive silver
stain, both of which bind tightly to
all proteins.
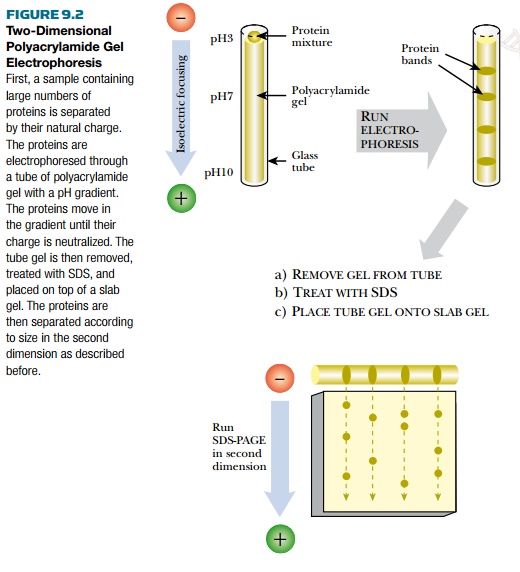
Thousands of proteins are found in cells, and many of these proteins are similar in size. However, resolving individual proteins into single bands on an acrylamide gel requires that the sample have only a few proteins, and these must be of different sizes. If the sample is from an entire cell, then the sample has so many proteins that individual bands will smear together. Using two-dimensional PAGE (2D-PAGE) can help alleviate this problem by first separating the proteins by their native charge in one dimension, then separating by size in a second dimension (Fig. 9.2). Isoelectric focusing is the term for separating proteins by their native charge. All proteins have an inherent natural charge due to the side chains of their amino acid residues. The total number of positively and negatively charged amino acids determines the natural charge on a protein. For separation by charge, the sample is loaded into the top of a gel with a pH gradient. When an electric field is applied, the proteins move along the pH gradient until their charge is neutralized. This step is usually done on a tube-shaped gel. After running, the gel is removed from its tube. The second dimension of 2D-PAGE is separation by size. The tube gel containing the separated proteins is treated with SDS in order to denature the proteins and coat them with negative charges as in regular PAGE. The tube gel is laid along the top of a polyacrylamide slab gel, and the proteins are separated by size. The gel is stained as described earlier.
2D-PAGE can resolve many proteins. Early studies on E. coli used 2D-PAGE to characterize all the proteins present under different conditions, about 1000 different proteins. Larger 2D-PAGE gels have been developed to study larger proteomes and can separate more than 10,000 different proteins into single spots. Identifying each spot on a 2D gel is a major task. Scientist interested in studying the proteome or translatome can stain the proteins and cut out each spot of interest. The protein trapped in each piece of gel can be digested into peptide fragments and identified by mass spectrometry (see later discussion). Another key feature of 2D-PAGE is the ability to quantify the relative amounts of different proteins. The size of the spot indicates the relative abundance of that protein. This can be quantified by scanning with a laser. Computer analysis then determines the density of the spot and relative abundance.
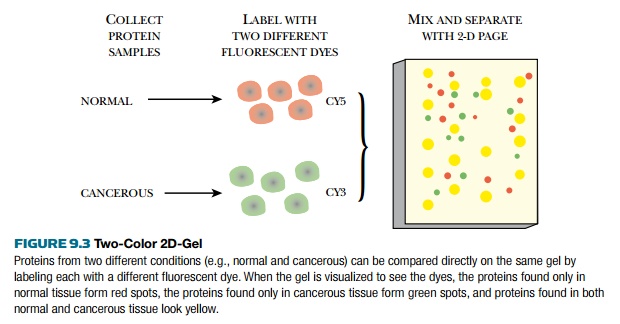
Two different samples can be
run together on the same gel to compare the proteins present under different
conditions (Fig. 9.3). Each sample is labeled with a different fluorescent dye.
Fluorescent labels such as methyl-Cy5 and propyl-Cy3 will fluoresce different
colors, but do not affect protein separation in 2D-PAGE. After both samples
have been run on the same gel, the dyes are activated by light. Where sample 1
has a protein not found in sample 2, the spot will fluoresce red, and when
sample 2 has a protein not found in sample 1, the spot will be green. When both
samples have the same amount of the same protein, the spot will be yellow. The
unique proteins can be isolated, digested, and identified using mass
spectroscopy (see later discussion).
Although 2D-PAGE has been
widely used, it does have some disadvantages. Certain classes of proteins are
underrepresented on a gel because they do not migrate through acrylamide.
Extremely large proteins often cannot enter into the gel matrix, whereas very
small proteins may travel off the end of the gel. Hydrophobic proteins may
travel through the gel, but their dynamics is altered because of their
hydrophobic surfaces. These proteins often run at different positions than
expected based on molecular weight. Proteins that are scarce within the cell,
such as transcription factors, are barely visible, if at all, with even the
most sensitive dyes. Abundant proteins, on the other hand, can distort the
location of nearby proteins within the gel because the gel becomes saturated at
that location. Another issue with 2D-PAGE is that proteins must be isolated
from the acrylamide for analysis by mass spectrometry (see later discussion).
Related Topics