Chapter: Biology of Disease: Toxicology
Paracetamol - Toxicology Poisons
COMMON POISONS
Humans are exposed to numerous xenobiotics, including drugs, pesticides, environmental pollutants, industrial chemicals and food additives. Any of these are potentially capable of perturbing the biochemistry and physiology of the body either directly or after being metabolically transformed. However, the most frequent poisons encountered in emergency toxicology include paracetamol (acetaminophen), aspirin, alcohols (ethanol, methanol, ethylene glycol), barbiturates (though this is now mainly historical), carbon monoxide, paraquat and several metals.
PARACETAMOL
Paracetamol (Figure 12.5) is used as an analgesic, that is, to relieve fever and pain. It is safe when taken at recommended doses but is toxic if overdosed. It is the commonest cause of admissions to hospital due to its wide availability. In the UK, overdoses cause approximately 150 deaths annually.
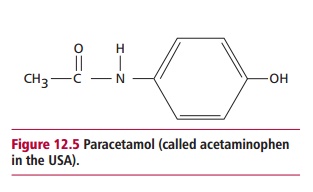
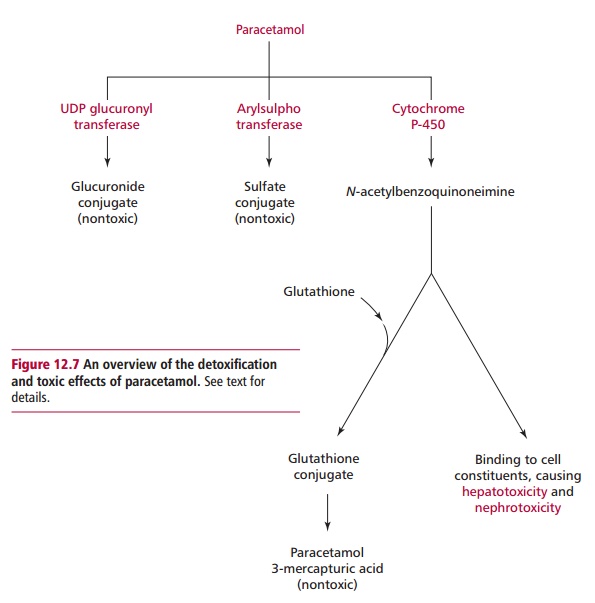
Paracetamol is rapidly absorbed from the stomach and upper GIT. The majority of ingested paracetamol is metabolized by conjugation with sulfate (~30%) or glucuronide (~60%) in the liver as described to form nontoxic metabolites. However, approximately 10% is metabolized by cytochrome P-450 to produce a highly reactive intermediate called N-acetyl-p-benzoquinoneimine (NABQI). It is possible for NABQI to be metabolized by conjugation with glutathione in hepatocytes to produce a nontoxic mercapturic acid. Glutathione is a tripeptide consisting of γ glutamate, cysteine and glycine residues, which will be further mentioned. It functions as a coenzyme in several oxidation– reduction reactions (Figure 12.6). When an overdose of paracetamol is ingested, liver detoxification systems may become saturated and the large amount of NABQI produced exhausts the limited stores of glutathione. As a consequence, NABQI binds to sulfhydryl groups of hepatocyte proteins, forming irreversible complexes that result in acute hepatic necrosis, that is, cell death (Figure 12.7). Paracetamol is also metabolized in cells of renal tubules and, in an overdose, renal tubular necrosis may also occur. In the presence of hepatic damage there is usually only a small amount of renal damage but occasionally this may be the major presenting feature of paracetamol poisoning. Alcohol in chronic alcoholics and the drugs phenobarbitone and phenytoin, used to treat epilepsy, may induce the synthesis of cytochrome P-450 enzymes and cause increased production of NABQI. As a consequence, hepatotoxicity may occur following a relatively small overdose of paracetamol in such patients. Severe toxicity is also more likely in people whose intracellular stores of glutathione are depleted as a result of starvation or protein malnutrition .
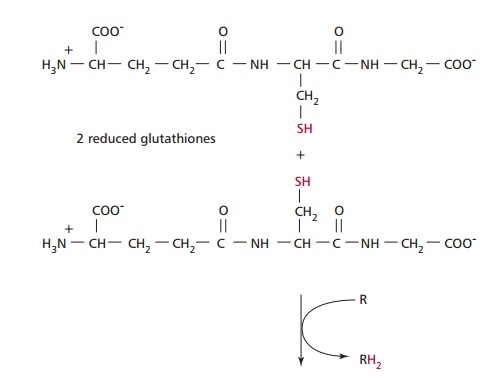
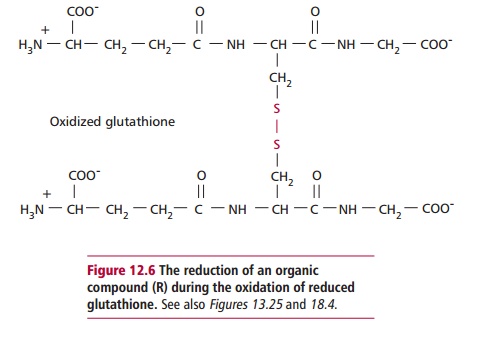
Substances, such as cimetidine, that inhibit the cytochrome P-450 system without interfering with glucuronidation or sulfation could potentially reduce paracetamol hepatotoxicity.
Toxic doses
A dose of 15 g for adults and 4 g for children is normally sufficient to cause hepatotoxicity. It has been suggested that this amount of paracetamol depletes liver glutathione concentrations by 70% in a 70 kg man. However, there is wide variation in the metabolic handling of paracetamol by the body and large overdoses of over 50 g have been known to have little effect in some patients. The incidence of hepatotoxicity in children is significantly lower at concentrations of paracetamol in blood that would be potentially toxic in adults. Animal studies have suggested that turnover of glutathione is age dependent, hence younger animals can tolerate higher doses of paracetamol.
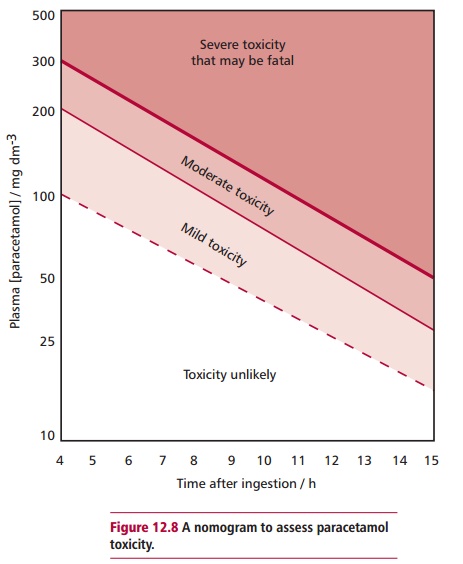
Often estimates of the amount of paracetamol ingested, for example by a potential suicide, are unreliable. Thus, predictions of hepatotoxicity should be made only on the basis of serum concentrations. In general, concentrations of paracetamol greater than 300 mg dm–3 cause serious liver damage 4 h after ingestion whereas values below 120 mg dm–3 show no toxicity.
Clinical features of paracetamol poisoning
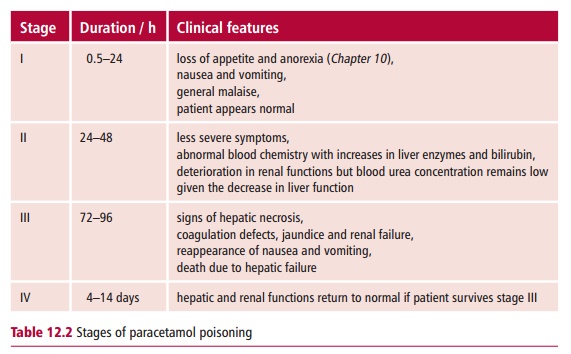
The signs and symptoms of paracetamol overdose are insidious, especially in the earlier stages. The clinical features can be divided into three phases, with a fourth occurring if the person survives toxicity and are described in Table12.2.
Laboratory investigations of paracetamol poisoning
Estimating the concentration of paracetamol in plasma is useful for assessing the probability of patients developing hepatotoxicity. A nomogram (Figure12.8) is available for paracetamol poisoning but should only be used whenthe size of the overdose and the approximate time of ingestion are known. Blood samples for paracetamol determination should be drawn at least 4 h postdose to allow for its complete absorption and the serum concentration to peak. The concentration of paracetamol in plasma can be used as a guide to patient management. Other tests that may be useful are determining the
activities of liver transaminases and measuring serum bilirubin to monitor liver functions and determining serum creatinine concentrations to assess renal function . Paracetamol poisoning causes an increased prothrombin time which is the time taken for blood clotting to occur in a sample of blood to which calcium and thromboplastin have been added.
Management of paracetamol poisoning
Antidotes to paracetamol poisoning include methionine and N-acetylcysteine. Both promote the synthesis of glutathione in the liver, increasing its capacity to detoxify the active metabolite. Methionine may be given orally at 2.5 g every 4 h for 12 h in early or uncomplicated cases, while N-acetylcysteine is administered parenterally and is more appropriate for patients who present late, or are comatose or vomiting. N-acetylcysteine can act as a glutathione substitute and enhances conjugation with sulfate (see earlier). It also limits liver damage by reducing inflammation and improving the microcirculation in the liver. Treatment gives maximal benefit if started within 10 h of ingestion although it may still be beneficial for up to 24–30 h. General procedures, such as administration of activated charcoal, can reduce gastrointestinal absorption of paracetamol if given in the first hour of an overdose. Gastric lavage may be used in patients who have ingested large amounts of paracetamol and present within an hour of ingestion.
Related Topics