Chapter: Biology of Disease: Toxicology
Drug Action, Metabolism, Distribution and Excretion
DRUG ACTION, METABOLISM,
DISTRIBUTION AND EXCRETION
Drugs are xenobiotics and may be defined as any
substance, other than food, that affects a living process. Pharmacology is the study of the effects of drugs in the
prevention, diagnosis, and treatment or cure of disease. Such drugs are often
referred to as medicines, which distinguishes them from other drugs that are
used for pleasure, such as some narcotics. Pharmacotherapeutics is that branch
of pharmacology concerned with the administration of drugs for prevention and
treatment of disease.
Drugs can be classified according to their chemical
structure but, more often, in terms of their pharmacological effects. For
example, they can be divided into three groups: chemotherapeutic drugs, which
are used to treat infectious diseases; pharmacodynamic drugs, such as sedatives
that are used in the treatment of noninfectious diseases; and a number of
miscellaneous agents including narcotics and analgesics.
Any single drug may have a chemical, brand and a
generic name. The chemical name is given according to the rules of chemical
nomenclature, whereas the brand name is given by the manufacturer. The generic
name is a common, established name given to a drug irrespective of that of its
manufacturer.
Most drugs act on cells to alter a biological
function. This pharmacological effect occurs as a consequence of the drug
reacting with a receptor that controls a particular function, or because the
drug alters a physiological mechanism which affects that function. For many
drugs, the extent and duration of the pharmacological effect are proportional
to the concentration of the drug at the receptor. The site at which the drug
acts to produce a pharmacological effect is called its site of action. The
mechanism of action of the drug is the biochemical or physiological process occurring
at the site of action to produce the pharmacological effect. Drug receptors
include enzymes and structural or transport proteins. However, some receptors
are nonprotein that bind to the drug to form a complex which alters the
permeability of the membranes or the transcription of DNA. Some drugs have a
structure similar to endogenous molecules and compete with them for binding
sites. Drugs may also act by preventing the formation, release, uptake or
transport of key substances in the body or by forming complexes with molecules
that can then activate receptors.
The binding of a drug to its receptor usually depends
on relatively weak forces, such as van der Waals forces and hydrogen and ionic
bonds and thus the formation of the drug–receptor complex that elicits the
response is normally freely reversible. Hence the response to any drug is not
permanent.
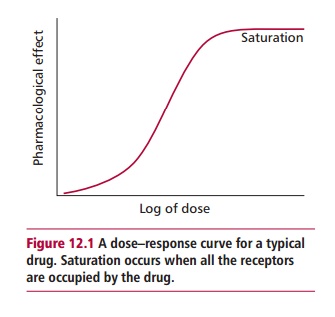
However, the response is dose dependent and, indeed,
a dose–response relationship exists between the concentration of drug in the
serum and the pharmacological effect. This response eventually reaches a
maximum effect because the receptor becomes saturated with the drug (Figure 12.1). The therapeutic range is
the concentrations of drug in the serum that is appropriate for therapy. The
dosage of any drug is planned to give a serum concentration within its
therapeutic range. Therapeutic drug monitoring is often necessary to determine
which given doses of a drug result in serum concentrations within the
therapeutic range. The serum concentration of the drug must not fall below its
minimum effective concentration (MEC) otherwise it will be ineffective.
However, neither should it rise above its minimum toxic concentration (MTC)
because of the danger of metabolic or structural damage. The time required for
the concentration of a drug in the blood to decline to half its original value
is referred to as its half-life (t1/2).
It is essential that a number of properties relating
to a medicinal drug, for example its pharmacodynamics and pharmacokinetics, are
first ascertained. Pharmacodynamics describes how the drug interacts with its
target site and the biochemical and physiological processes that result in any
therapeutic or toxic effects. Pharmacokinetics relates to the uptake,
distribution, metabolism and excretion from the body.
Most drugs are given orally for convenience, although
they can be administered intravenously, intramuscularly or subcutaneously. When
given orally, the absorption of the drug depends on its ability to disassociate
from its dosing form, dissolve in gastrointestinal fluids and diffuse across
the gut wall into the blood. The rate and extent of drug absorption varies with
the nature of the drug, the matrix in which it is dissolved and the region of
the gastrointestinal tract (GIT) where it is absorbed. The proportion of the
drug absorbed into the circulation is referred to as its bioavailability. For
an orally delivered drug, this should generally be greater than 70% to be of
therapeutic use. However, when the site of action is the GIT lumen itself, for
example treating a GIT infection, then a low bioavailability would be
advantageous.
A number of drugs undergo what is referred to as
first pass metabolism. They are absorbed rapidly and completely by the GIT but,
nevertheless, have low bioavailability because they are transported to the
liver in the hepatic portal vein and metabolized and have not entered the
systemic circulation. Drugs with delayed absorption are sometimes required and
special slow or sustained release formulations have been developed for these
cases. Such drugs can be taken orally at less frequent intervals. Certain
diseases that affect the GIT and the interaction of some drugs and foods in the
GIT can delay their absorption.
Following absorption, drug distribution occurs when
the compound enters the vascular system. The physical, chemical and molecular
properties of the drug can influence its distribution. Its distribution may
also be influenced by its binding to blood components and receptors and its
ability to dissolve in lipids and pass through biological membranes. Many drugs
bind to plasma proteins and often an equilibrium is established between
protein-bound and free drug. Only the free fraction is able to interact with
receptors or cross cellular membranes. Any factor that changes drug– protein
interactions may alter the distribution, pharmacological effects and excretion
of the drug.
Related Topics