Chapter: Essential Anesthesia From Science to Practice : Applied physiology and pharmacology : A brief pharmacology related to anesthesia
Neuromuscular blockers and their antagonists
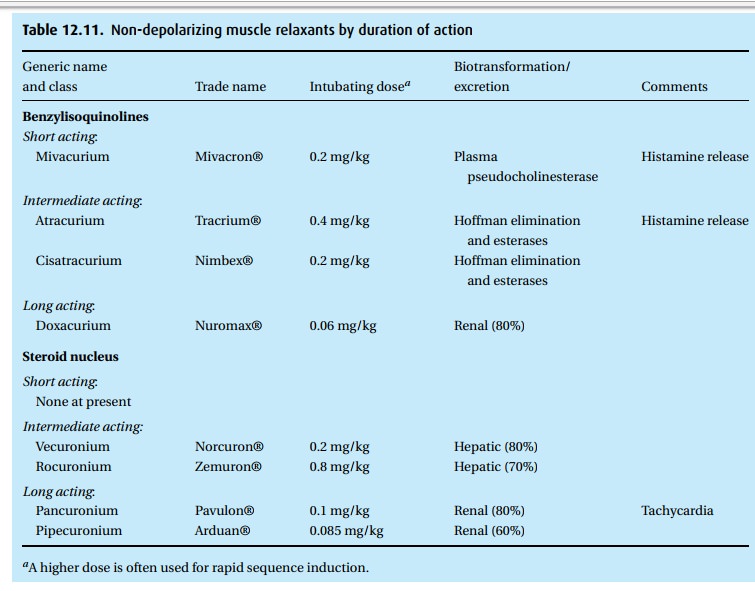
Neuromuscular blockers and their
antagonists (Table 12.11)
Even
though the title presents the official name, we will call them muscle
relax-ants with the understanding that we are talking about drugs used in
anesthesia to facilitate tracheal intubation and to ease the surgeon’s work.
The good news about muscle relaxants is that they affect only striated,
voluntary muscles, but not the myocardium and the smooth muscles under
autonomic control (including the uterus). Being quaternary ammonium compounds,
all muscle relaxants carry a charge and thus do not readily cross the
blood–brain barrier (no effect on the brain) or the placenta (no effect on the
fetus). The bad news is that the relax-ants do not spare the muscles of
ventilation. That fact has cost many lives when partially paralyzed patients
became hypoxemic because inadequate ventilation was allowed to persist during
and particularly after anesthesia. Do not forget that muscle relaxants have no
anesthetic effect, that a patient paralyzed by mus-cle relaxants has no way of
signaling that he is in pain, uncomfortable or short of breath, a fact not lost
on those patients suffering intra-operative awareness. There
Note also that even the pharmacological reversal of
the effect of muscle relaxants has undesirable side effects. Whenever muscle
relax-ants are used, we assume great responsibility for the safety of the
patient. Many procedures do not require muscle relaxants. When no muscle
relaxants are used, the patient can breathe spontaneously, which they tend to
do very well indeed as long as we are not heavy handed with CNS depressants.
Muscle relaxants are usually divided into depolarizing and non-depolarizing
drugs.
Depolarizing muscle relaxants
Succinylcholine
(Anectine®) is the only depolarizing drug still in use. It has been around for
50 years and has served us well because of two characteristics: it is rapid in
onset and short in duration, being hydrolyzed by plasma cholinesterases.
Indeed,
perhaps as much as 90% of the intravenously injected drug is hydrolyzed before
reaching the effector site at the neuromuscular junction. Patients defi-cient
in plasma cholinesterase will be paralyzed for several hours from a standard
intubating dose of 1 mg/kg, which should last for only 5 minutes or so.
Cholinesterase
deficiency can be genetic or acquired. One in 3200 patients (less often in
Oriental and African peoples) may be homozygous for atypical cholinesterase.
When we suspect this because of a family history or a previous anesthetic
complication, we can test the patient’s plasma in vitro, using dibucaine
(Nupercaine®), a local anesthetic. Dibucaine strongly (80%) inhibits normal or
‘typical’ plasma cholinesterase but not the atypical cholinesterase (20%). A
report of a ‘dibucaine number’ of 80 is good news, suggesting that the patient
is homozy-gous for typical plasma cholinesterase. A dibucaine number of 20 or
so would be found in a patient homozygous for atypical plasma cholinesterase,
who would have an abnormally protracted effect from succinylcholine. Dibucaine
numbers between these extremes suggest a heterozygous genetic make-up. In the
patient heterozygous for normal plasma cholinesterase, the succinylcholine
effect is likely to be doubled or tripled (5 to 15 minutes). Incidentally,
patients homozygous for atypical cholinesterase are quite asymptomatic – as
long as no one gives them suc-cinylcholine or other drugs dependent on
hydrolysis by plasma cholinesterases. We see the acquired deficiency –
characterized by decreased blood levels of nor-mal plasma cholinesterase – in
patients exposed to organophosphates (chemical warfare and pesticides) and
those on echothiophate (for glaucoma) who would also more slowly break down
some other esters such as local anesthetics of the ester type.
Succinylcholine
does not compete with acetylcholine at the neuromuscular junction; instead, it
depolarizes the muscle and in so doing, it opens ion channels, much like
acetylcholine does, but the channels stay open much longer. Potassium begins to
leak out and serum potassium levels can rise by 0.5 mEq/L after an intubating
dose (succinylcholine 1 mg/kg). In damaged (crush or burn injuries) or
degenerating muscles (after spinal cord injury or in muscular dystrophy), this
potassium leakage can be exaggerated to the point where the cardiac effects of
hyperkalemia become life-threatening. The risk of yet unrecognized muscular
dystrophy, together with the potential for a bradycardic response, has limited
the use of succinylcholine in children. Succinylcholine has several additional
undesirable properties. Before paralysis sets in, it causes fasciculation of
striated muscle, a feature that has been blamed for post-operative myalgia
experienced by some patients and for a transient rise in intragastric and
intracranial pressures. By a mechanism not well understood, intra-ocular pressure
also rises briefly after an intubating dose. Therefore, we do not use the drug
in patients with an open eye lest the patient lose vitreous. In the past,
succinylcholine was often used as a continuous infusion. In that application,
it loses its advantage of a short-acting depolarizing blocker because the
patient will develop a so-called phase II block that looks as if the patient
had been given a non-depolarizing muscle relaxant (see chapter on Monitoring).
When
tracheal intubation fails and the succinylcholine effect wears off, we might be
tempted to administer a second dose of succinylcholine within a few minutes of
the first dose. This is dangerous, possibly causing severe bradycardia and even
asystole presumably triggered by cholinergic effects of the second dose.
Therefore, always administer i.v. atropine or glycopyrrolate (0.6 mg or 0.4 mg,
respectively, for the average adult) before giving a second dose of
succinylcholine.
Non-depolarizing muscle relaxants
The
South American Indians did not know that they were delivering a
non-depolarizing drug in their blowpipes when hunting monkeys. We might wonder
if they were astonished that they were not weakened or paralyzed when eating
the curare-poisoned monkey meat. Being quaternary, bulky molecules, D-tubocurare
is not absorbed from the gut. Today, we have a long list of non-depolarizing
mus-cle relaxants, which act by competing with acetylcholine at the
neuromuscular endplate. They are either benzylisoquinolines (like the original
D-tubocurare) or steroid derivatives. We can roughly classify them as
short-acting, i.e., less than 30 minutes, intermediate-acting (between 30 and
60 minutes), and long-acting (over 1 hour). The duration is affected by the
dose and by how we define dura-tion. For example, an intubating dose (a lot of
relaxation!) of a short-acting drug might provide adequate surgical relaxation
(soft abdominal muscles) for 1/2 hour; however, after
these 30 minutes, the patient might not be capable of maintaining normal blood
gases without assisted ventilation. Table 12.10provides
a short list of some of the currently used drugs with certain of their
characteristics. For each drug we give an “intubating dose.”
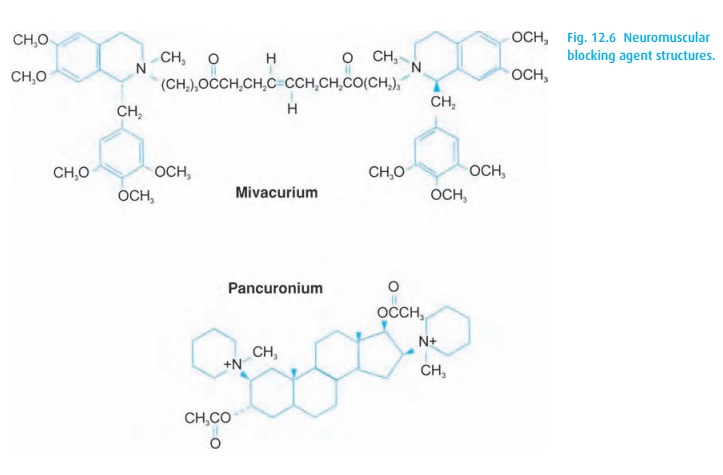
In Fig. 12.6we show mivacurium (Mivacron®) representing
the benzyliso-quinolines and pancuronium (Pavulon®) for its steroid nucleus.
Observe the ester linkage in mivacurium, which can be attacked by
cholinesterases, making it a short-acting drug; however, subject to prolonged
effect with plasma cholin-esterase deficiencies.
Muscle relaxant reversal
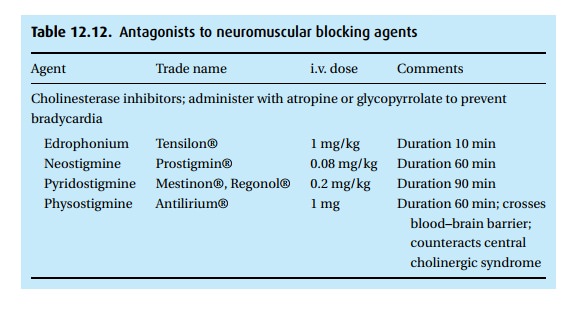
We do not reverse the effect of succinylcholine with an antagonist. Instead, we unwearingly ventilate the patient’s lungs until the block has worn off, even if that takes hours in a patient homozygous for atypical cholinesterase. This differs from the non-depolarizing drugs. An excess of acetylcholine, the physiologic transmit-ter substance at the endplate, will compete with the non-depolarizing relaxant for access to the endplate. Thus we give a cholinesterase inhibitor, prolonging the life of acetylcholine so it can better compete. Because these inhibitors act not only on the neuromuscular apparatus but also generate an excess of acetylcholine at autonomic sites, we add an anticholinergic drug that acts primarily on the auto-nomic (muscarinic) receptors. Thus, atropine or glycopyrrolate (Robinul®) can prevent the unwanted autonomic effects of the cholinesterase inhibitors, such as excessive salivation, bradycardia and intestinal cramping.
The most
commonly used cholinesterase inhibitors are neostigmine (Prostigmin®) and
edrophonium (Tensilon®). Both are quaternary ammonium compounds that do not
cross the blood–brain barrier, and both are potent cholinesterase inhibitors.
While they show small differences in their action, either one can serve when
the weakening effect of a muscle relaxant must be reversed. Neostigmine takes
up to 10 minutes after an intravenous dose to reach its peak effect;
edrophonium is much faster. Reversal of neuromuscular blockade cannot be
achieved unless a few receptors are unblocked to give acetylcholine a fighting
chance. Using a “twitch monitor” (see Monitoring), we do not administer
reversal agents until we detect at least a small response to stimulation
(indicating that no more than 90% of the receptors are blocked). Typical
reversal doses are:
neostigmine
up to 0.08 mg/kg or edrophonium up to 1 mg/kg with
atropine
or glycopyrrolate up to 15 mcg/kg.
These
doses must be adjusted to meet the patient’s requirements (see Table 12.12).
A new
category of drugs, the cyclodextrins, now in clinical trials, might offer
advantages. They appear to chelate the muscle relaxants without antagonizing
them via the inhibition of cholinesterases.
The
Monitoring chapter details assessment of neuromuscular blockade and muscle
strength.
Dantrolene
Dantrolene
(Dantrium®) finds use as an oral medication in the treatment of muscle spasms
in multiple sclerosis, cerebral palsy, stroke, or injury to the spine. It
affects skeletal muscles directly, i.e., beyond the neuromuscular junction. In
the treatment of malignant hyperthermia, we count on its ability to
re-establish a normal level of the dangerously elevated ionized calcium in the
myoplasm. We start with a bolus of 1–2 mg/kg, repeated every 5–10 minutes as
necessary, to a maximum of 10 mg/kg. The drug comes in vials containing 20 mg
dantrolene and 3000 mg mannitol. This has to be dissolved with 60 ml sterile
water. To administer 2–3 mg/kg to an adult will require many vials and an extra
pair of hands to prepare and administer the drug.
Related Topics