Chapter: Basic & Clinical Pharmacology : Agents Used in Cardiac Arrhythmias
Mechanisms of Arrhythmias
MECHANISMS OF ARRHYTHMIAS
Many
factors can precipitate or exacerbate arrhythmias: ischemia, hypoxia, acidosis
or alkalosis, electrolyte abnormalities, excessive catecholamine exposure,
autonomic influences, drug toxicity (eg, digitalis or antiarrhythmic drugs),
overstretching of cardiac fibers, and the presence of scarred or otherwise
diseased tissue. However, all arrhythmias result from (1) disturbances in
impulse formation, (2) disturbances in impulse conduction, or (3) both.
Disturbances of Impulse Formation
The
interval between depolarizations of a pacemaker cell is the sum of the duration
of the action potential and the duration of the diastolic interval. Shortening
of either duration results in an increase in pacemaker rate. The more important
of the two, dia-stolic interval, is determined primarily by the slope of phase
4 depolarization (pacemaker potential). Vagal discharge and β-receptor–blocking
drugs slow normal pacemaker rate by reduc-ing the phase 4 slope (acetylcholine
also makes the maximum diastolic potential more negative). Acceleration of
pacemaker dis-charge is often brought about by increased phase 4 depolarization
slope, which can be caused by hypokalemia, β-adrenoceptor stimulation, positive
chronotropic drugs, fiber stretch, acidosis, and partial depolarization by
currents of injury.
Latent
pacemakers (cells that show slow phase 4 depolarization even under normal
conditions, eg, some Purkinje fibers) are par-ticularly prone to acceleration
by the above mechanisms. However, all cardiac cells, including normally
quiescent atrial and ventricu-lar cells, may show repetitive pacemaker activity
when depolarized under appropriate conditions, especially if hypokalemia is
also present.
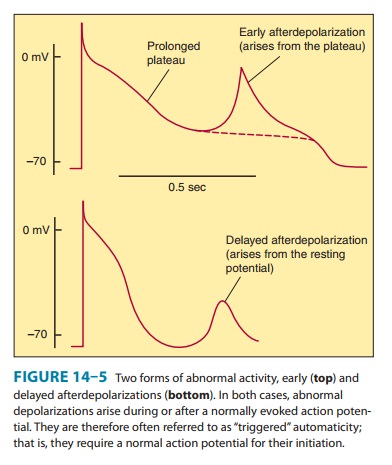
Afterdepolarizations
(Figure 14–5) are transient depolariza-tions that interrupt phase 3 (early afterdepolarizations, EADs) or
phase 4 (delayed afterdepolarizations,
DADs). EADs are usu-ally
exacerbated at slow heart rates and
are thought to contribute to the development of long QT-related arrhythmias
(see Box:Molecular& Genetic Basis of Cardiac Arrhythmias). DADs, on the
other hand, often occur when intracellular calcium is increased . They are
exacerbated by fast heart rates and
are thought to be responsible for some arrhythmias related to digitalis excess,
to catecholamines, and to myocardial ischemia.
Disturbances of Impulse Conduction
Severely
depressed conduction may result in simple block,
eg, AV nodal block or bundle branch block. Because parasympathetic con-trol of
AV conduction is significant, partial AV block is sometimes relieved by
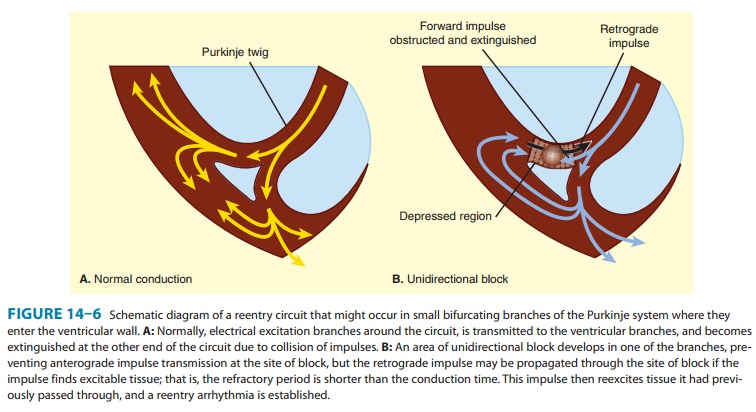
atropine. Another common abnormality of conduction is reentry (also known as “circus movement”), in which one impulse
reenters and excites areas of the heart more than once (Figure 14–6).The path
of the reentering impulse may be confined to very small areas, eg, within or
near the AV node, or it may involve large portions of the atrial or ventricular
walls. Some forms of reentry are strictly anatomically determined; for example,
in Wolff-Parkinson-White syndrome, the reentry circuit consists of atrial
tissue, the AV node, ventricular tissue, and an accessory AV connection (bundle
of Kent, a bypass tract). In other cases (eg, atrial or ventricular
fibrillation),
multiple
reentry circuits, determined by the varying properties of the cardiac tissue,
may meander through the heart in apparently random paths. The circulating
impulse often gives off “daughter impulses” that can spread to the rest of the
heart. Depending on how many round trips through the pathway the reentrant
impulse makes before dying out, the arrhythmia may be manifest as one or a few
extra beats or as a sustained tachycardia.
For
reentry to occur, three conditions must coexist, as indi-cated in Figure 14–6.
(1) There must be an obstacle (anatomic or physiologic) to homogeneous
conduction, thus establishing a circuit around which the reentrant wavefront
can propagate.
There must be unidirectional block at some point in the circuit;
that is, conduction must die out in one direction but continue in the opposite
direction (as shown in Figure 14–6, the impulse can gradually decrease as it
invades progressively more depolarized tissue until it finally blocks—a process
known as dec-remental conduction). (3) Conduction time around the circuit must
be long enough that the retrograde impulse does not enter refractory tissue as
it travels around the obstacle; that is, the con-duction time must exceed the
effective refractory period. It is important to note that reentry depends on
conduction that has been depressed by some critical amount, usually as a result
of injury or ischemia. If conduction velocity is too slow, bidirectional block
rather than unidirectional block occurs; if the reentering impulse is too weak,
conduction may fail, or the impulse may arrive so late that it collides with
the next regular impulse. On the other hand, if conduction is too rapid—ie,
almost normal—bidi-rectional conduction rather than unidirectional block will
occur. Even in the presence of unidirectional block, if the impulse travels around
the obstacle too rapidly, it will reach tissue that is still refractory.
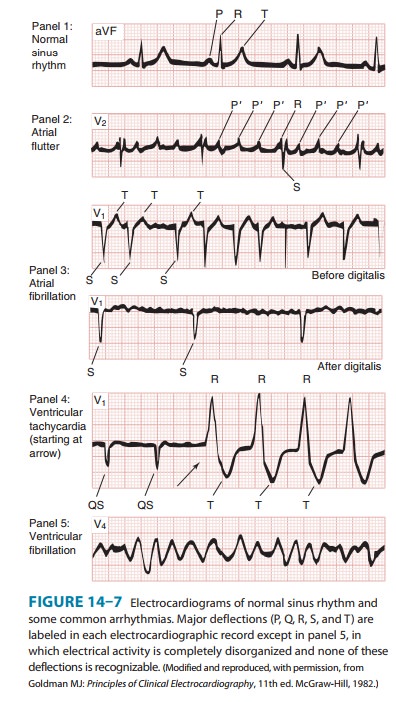
Representative electrocardiograms of important arrhyth-mias are shown in
Figures 14–7 and 14–8.
Slowing of conduction may be due to depression of sodium current, depression of calcium current (the latter especially in the AV node), or both. Drugs that abolish reentry usually work by further
slowing depressed conduction (by blocking the sodium or calcium current) and
causing bidirectional block. In theory, accel-erating conduction (by increasing
sodium or calcium current) would also be effective, but only under unusual
circumstances does this mechanism explain the action of any available
drug.Lengthening (or shortening) of the refractory period may also make reentry
less likely. The longer the refractory period in tissue near the site of block,
the greater the chance that the tissue will still be refractory when reentry is
attempted. (Alternatively, the shorter the refractory period in the depressed
region, the less likely it is that unidirectional block will occur.) Thus,
increased disper-sion of refractoriness is one contributor to reentry, and
drugs may suppress arrhythmias by reducing such dispersion.
Molecular & Genetic Basis of Cardiac Arrhythmias
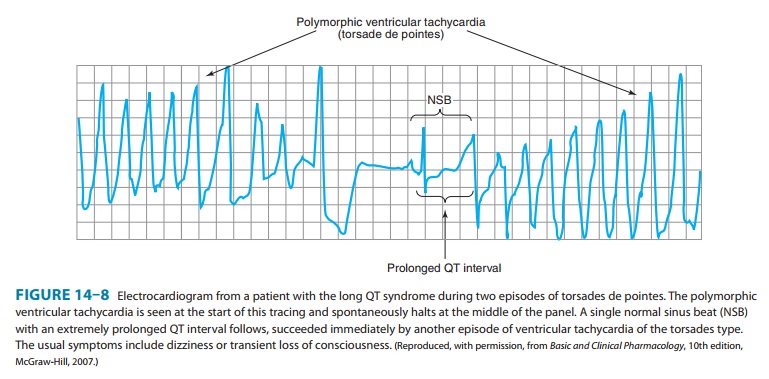
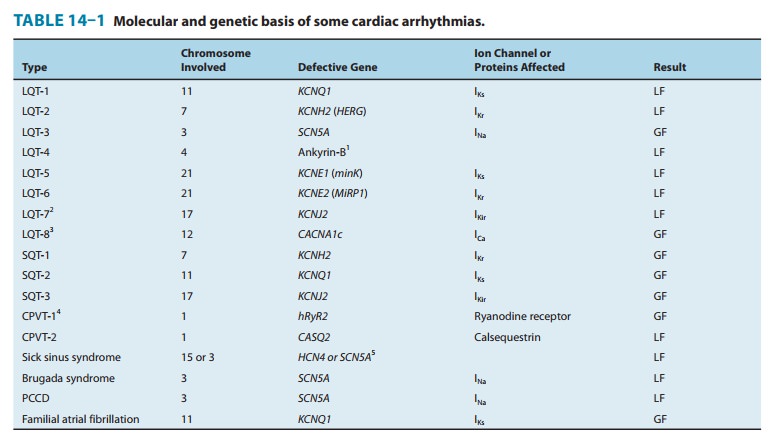
It is now possible to define the molecular basis of several congenital and acquired cardiac arrhythmias. The best example is the polymor-phic ventricular tachycardia known as torsades de pointes (shown in Figure 14–8), which is associated with prolongation of the QT interval (especially at the onset of the tachycardia), syncope, and sudden death. This must represent prolongation of the action potential of at least some ventricular cells (Figure 14–1). The effect can, in theory, be attributed to either increased inward current (gain of function) or decreased outward current (loss of function) during the plateau of the action potential. In fact, recent molecular genetic studies have identified up to 300 different mutations in at least eight ion channel genes that produce the congenital long QT (LQT) syndrome (Table 14–1), and different mutations may have different clinical implications. Loss-of-function mutations in potassium channel genes produce decreases in outward repolarizing current and are respon-sible for LQT subtypes 1, 2, 5, 6, and 7. HERG and KCNE2 (MiRP1) genes encode subunits of the rapid delayed rectifier potassium current (IKr), whereas KCNQ1 and KCNE1 (minK) encode subunits of the slow delayed rectifier potassium current (IKs). KCNJ2 encodes an inwardly rectifying potassium current (IKir). In contrast, gain-of-function muta-tions in the sodium channel gene (SCN5A) or calcium channel gene (CACNA1c) cause increases in inward plateau current and are respon-sible for LQT subtypes 3 and 8, respectively.Molecular genetic studies have identified the reason why con-genital and acquired cases of torsades de pointes can be so strik-ingly similar. The potassium channel IKr (encoded by HERG) is blocked or modified by many drugs (eg, quinidine, sotalol) or elec-trolyte abnormalities (hypokalemia, hypomagnesemia, hypocalce-mia) that also produce torsades de pointes. Thus, the identification of the precise molecular mechanisms underlying various forms ofthe LQT syndromes now raises the possibility that specific therapies may be developed for individuals with defined molecular abnor-malities. Indeed, preliminary reports suggest that the sodium chan-nel blocker mexiletine can correct the clinical manifestations of congenital LQT subtype 3 syndrome. It is likely that torsades de pointes originates from triggered upstrokes arising from early after-depolarizations (Figure 14–5). Thus, therapy is directed at correcting hypokalemia, eliminating triggered upstrokes (eg, by using β block-ers or magnesium), or shortening the action potential (eg, by increasing heart rate with isoproterenol or pacing)—or all of these.The molecular basis of several other congenital cardiac arrhyth-mias associated with sudden death has also recently been identi-fied. Three forms of short QT syndrome have been identified that are linked to gain-of-function mutations in three different potas-sium channel genes (KCNH2, KCNQ1, and KCNJ2). Catecholaminergic polymorphic ventricular tachycardia, a disease that is characterized by stress- or emotion-induced syncope, can be caused by genetic mutations in two different proteins in the sarcoplasmic reticulum that control intracellular calcium homeostasis. Mutations in two different ion channel genes (HCN4 and SCN5A) have been linked to congenital forms of sick sinus syndrome. The Brugada syndrome, which is characterized by ventricular fibrillation associated with persistent ST-segment elevation, and progressive cardiac conduc-tion disorder (PCCD), characterized by impaired conduction in the His-Purkinje system and right or left bundle block leading to com-plete atrioventricular block, have both been linked to several loss-of-function mutations in the sodium channel gene, SCN5A. At least one form of familial atrial fibrillation is caused by a gain-of-function mutation in the potassium channel gene, KCNQ1
Related Topics