Chapter: Basic & Clinical Pharmacology : Agents Used in Cardiac Arrhythmias
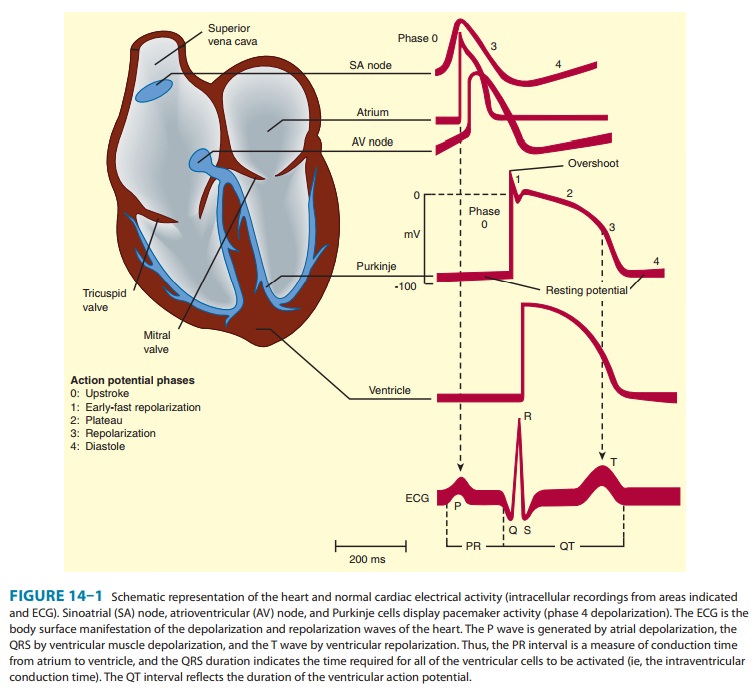
Electrophysiology of Normal Cardiac Rhythm
ELECTROPHYSIOLOGY OF NORMAL
CARDIAC RHYTHM
The
electrical impulse that triggers a normal cardiac contrac-tion originates at
regular inter vals in the sinoatrial (SA) node ( Figure 14ÔÇô1), usually at a
frequency of 60ÔÇô100 bpm. This impulse spreads rapidly through the atria and
enters the atrioventricular (AV) node, which is normally the only con-duction
pathway between the atria and ventricles. Conduction through the AV node is
slow, requiring about 0.15 seconds. (This delay provides time for atrial contraction
to propel blood into the ventricles.) The impulse then propagates over the
His-Purkinje system and invades all parts of the ventri-cles, beginning with
the endocardial surface near the apex and ending with the epicardial surface at
the base of the heart. Ventricular activation is complete in less than 0.1
seconds; therefore, contraction of all of the ventricular muscle is nor-mally
synchronous and hemodynamically effective.
Arrhythmias consist of
cardiac depolarizations that deviate from the above description in one or more
aspects: there is an abnormality in the site of origin of the impulse, its rate
or regularity, or its conduction.
Ionic Basis of Membrane
Electrical Activity
The
transmembrane potential of cardiac cells is determined by the concentrations of
several ionsÔÇöchiefly sodium (Na+), potassium (K+), calcium (Ca2+), and chloride (ClÔÇô)ÔÇöon
either side of the mem-brane and the permeability of the membrane to each ion.
These water-soluble ions are unable to freely diffuse across the lipid cell
membrane in response to their electrical and concentration gradi-ents; they
require aqueous channels (specific pore-forming proteins) for such diffusion.
Thus, ions move across cell membranes in response to their gradients only at
specific times during the cardiac cycle when these ion channels are open. The
movements of the ions produce currents that form the basis of the cardiac
action potential. Individual channels are relatively ion-specific, and the flux
of ions through them is controlled by ÔÇ£gatesÔÇØ (flexible portions of thepeptide
chains that make up the channel proteins). Each type of channel has its own
type of gate (sodium, calcium, and some potas-sium channels are each thought to
have two types of gates). The channels primarily responsible for the cardiac
action potential (sodium, calcium, and several potassium) are opened and closed
(ÔÇ£gatedÔÇØ) by voltage changes across the cell membrane; that is, they are
voltage-sensitive. Most are also modulated by ion concentra-tions and metabolic
conditions, and some potassium channels are primarily ligand- rather than
voltage-gated.
All
the ionic currents that are currently thought to contribute to the cardiac
action potential are illustrated in Figure 14ÔÇô2. At rest, most cells are not
significantly permeable to sodium, but at the start of each action potential,
they become quite permeable . In electrophysiologic terms, the conductance of
the fast sodium
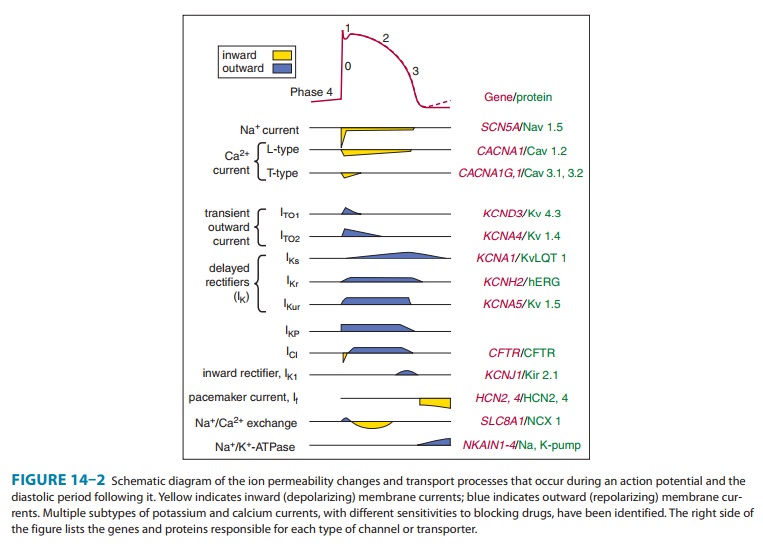
Similarly, calcium
enters and potassium leaves the cell with each action potential. Therefore, in
addition to ion channels, the cell must have mechanisms to maintain stable
transmembrane ionic condi-tions by establishing and maintaining ion gradients.
The most important of these active mechanisms is the sodium pump, Na+/ K+-ATPase. This pump and
other active ion carriers contribute indirectly to the transmembrane potential
by maintaining the gradients necessary for diffusion through channels. In
addition, some pumps and exchangers produce net current flow (eg, by exchanging
three Na+ for two K+ ions) and hence are termed ÔÇ£electrogenic.ÔÇØ
When
the cardiac cell membrane becomes permeable to a spe-cific ion (ie, when the
channels selective for that ion are open), movement of that ion across the cell
membrane is determined by OhmÔÇÖs law: current = voltage ├À resistance, or current = voltage ├ù conductance.
Conductance is determined by the properties of the individual ion channel
protein. The voltage term is the difference between the actual membrane
potential and the reversal potential for that ion (the membrane potential at
which no current would flow even if channels were open). For example, in the
case of sodium in a cardiac cell at rest, there is a substantial concentration
gradient (140 mmol/L Na+ outside; 10ÔÇô15 mmol/L Na+ inside) and an
electrical gradient (0 mV outside; 90 mV inside) thatwould drive Na+ into cells. Sodium
does not enter the cell at rest because sodium channels are closed; when sodium
channels open, the very large influx of Na+ accounts for phase 0
depolarization of the action potential. The situation for K+ in the resting
cardiac cell is quite different. Here, the concentration gradient (140 mmol/L
inside; 4 mmol/L outside) would drive the ion out of the cell, but the
electrical gradient would drive it in; that is, the inward gradi-ent is in
equilibrium with the outward gradient. In fact, certain potassium channels
(ÔÇ£inward rectifierÔÇØ channels) are open in the resting cell, but little current
flows through them because of this balance. The equilibrium, or reversal potential, for ions is
deter-mined by the Nernst equation:
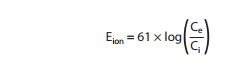
where
Ce and Ci are the extracellular and intracellular
concentra-tions, respectively, multiplied by their activity coefficients. Note
that raising extracellular potassium makes EK less negative. When
this occurs, the membrane depolarizes until the new EK is reached.
Thus, extracellular potassium concentration and inward rectifier channel
function are the major factors determining the membrane potential of the
resting cardiac cell. The conditions required for application of the Nernst
equation are approximated at the peak of the overshoot (using sodium
concentrations) and during rest (using potassium concentrations) in most
nonpacemaker cardiac cells. If the permeability is significant for both
potassium and sodium, the Nernst equation is not a good predictor of membrane
potential, but the Goldman-Hodgkin-Katz
equation may be used:
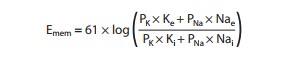
In
pacemaker cells (whether normal or ectopic), spontaneous depolarization (the
pacemaker potential) occurs during diastole (phase 4, Figure 14ÔÇô1). This
depolarization results from a gradual increase of depolarizing current through
special hyperpolarization-activated ion channels (If, also called Ih)
in pacemaker cells. The effect of changing extracellular potassium is more
complex in a pacemaker cell than it is in a nonpacemaker cell because the
effect on permeability to potassium is much more important in a pace-maker (see
Box: Effects of Potassium). In a pacemakerÔÇöespecially an ectopic oneÔÇöthe end
result of an increase in extracellular potassium is usually to slow or stop the
pacemaker. Conversely, hypokalemia often facilitates ectopic pacemakers.
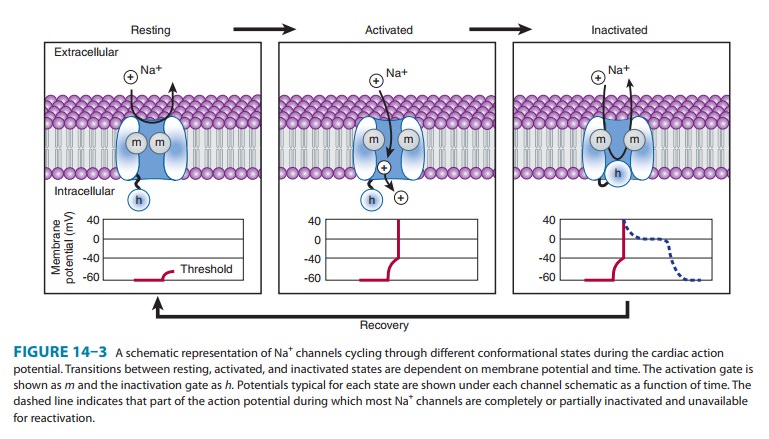
The Active Cell Membrane
In
normal atrial, Purkinje, and ventricular cells, the action potential upstroke
(phase 0) is dependent on sodium current. From a functional point of view, it
is convenient to describe the behavior of the sodium current in terms of three
channel states (Figure 14ÔÇô3). The cardiac sodium channel protein has been cloned,
and it is now recognized that these channel states actu-ally represent
different protein conformations. In addition, regions of the protein that
confer specific behaviors, such as volt-age sensing, pore formation, and
inactivation, are now being identified. The gates described below and in Figure
14ÔÇô3 repre-sent such regions.
Effects of Potassium
The effects of changes in serum potassium on cardiac action potential duration, pacemaker rate, and arrhythmias can appear somewhat paradoxical if changes are predicted based solely on a consideration of changes in the potassium electro-chemical gradient. In the heart, however, changes in serumpotassium concentration have the additional effect of alter-ing potassium conductance (increased extracellular potas-sium increases potassium conductance) independent of simple changes in electrochemical driving force, and this effect often predominates. As a result, the actual observed effects of hyperkalemia include reduced action potential duration, slowed conduction, decreased pacemaker rate, and decreased pacemaker arrhythmogenesis. Conversely, the actual observed effects of hypokalemia include prolonged action potential duration, increased pacemaker rate, and increased pacemaker arrhythmogenesis. Furthermore, pace-maker rate and arrhythmias involving ectopic pacemaker cells appear to be more sensitive to changes in serum potas-sium concentration, compared with cells of the sinoatrial node. These effects of serum potassium on the heart probably contribute to the observed increased sensitivity to potassium channel-blocking antiarrhythmic agents (quinidine or sotalol) during hypokalemia, eg, accentuated action potential prolon-gation and tendency to cause torsades de pointes.
Depolarization
to the threshold voltage results in opening of the activation (m) gates of sodium channels (Figure 14ÔÇô3,
middle). If the inactivation (h)
gates of these channels have not already closed, the channels are now open or
activated, and sodium permeability is markedly increased, greatly exceed-ing
the permeability for any other ion. Extracellular sodium therefore diffuses
down its electrochemical gradient into the cell, and the membrane potential
very rapidly approaches the sodium equilibrium potential, ENa (about
+70 mV when Nae= 140 mmol/L and Nai= 10 mmol/L). This
intense sodium current is very brief because opening of the m gates upon depolarization is promptly
followed by closure of the h gates
and inactivation of the sodium channels (Figure 14ÔÇô3, right).
Most
calcium channels become activated and inactivated in what appears to be the
same way as sodium channels, but in the case of the most common type of cardiac
calcium channel (the ÔÇ£LÔÇØ type), the transitions occur more slowly and at more
positive potentials. The action potential plateau (phases 1 and 2) reflects the
turning off of most of the sodium current, the waxing and waning of calcium
current, and the slow development of a repolar-izing potassium current.
Final
repolarization (phase 3) of the action potential results from completion of
sodium and calcium channel inactivation and the growth of potassium
permeability, so that the membrane potential once again approaches the
potassium equilibrium poten-tial. The major potassium currents involved in
phase 3 repolariza-tion include a rapidly activating potassium current (IKr)
and a slowly activating potassium current (IKs). These two potassium
cur-rents are sometimes discussed together as ÔÇ£IK.ÔÇØ It is noteworthy
that a different potassium current, distinct from IKr and IKs,
may control repolarization in SA nodal cells. This explains why some drugs that
block either IKr or IKs may prolong repolarization in
Purkinje and ventricular cells, but have little effect on SA nodal
repolarization (see Box: Molecular & Genetic Basis of Cardiac Arrhythmias).
The Effect of Resting Potential on Action Potentials
A
key factor in the pathophysiology of arrhythmias and the actions of
antiarrhythmic drugs is the relation between the resting potential of a cell
and the action potentials that can be evoked in it (Figure 14ÔÇô4, left panel).
Because the inactivation gates of sodium channels in the resting membrane close
over the potential range 75 to 55 mV, fewer sodium channels are available
for diffusion of sodium ions when an action potential is evoked from a resting
potential of 60 mV than when it is evoked from a resting potential of 80 mV.
Important consequences of the reduction in peak sodium permeability include
reduced maximum upstroke velocity (called Vmax, for maximum rate of
change of membrane voltage), reduced action potential amplitude, reduced
excitability, and reduced conduction velocity.
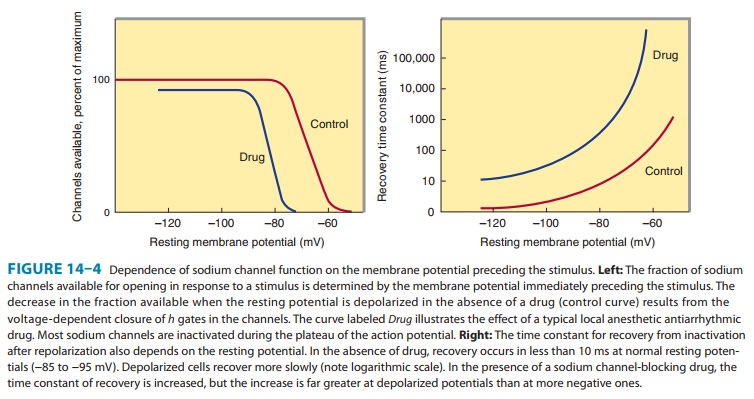
During
the plateau of the action potential, most sodium chan-nels are inactivated.
Upon repolarization, recovery from inactiva-tion takes place (in the
terminology of Figure 14ÔÇô3, the h
gates reopen), making the channels again available for excitation. The time
between phase 0 and sufficient recovery of sodium channels in phase 3 to permit
a new propagated response to an external stimulus is the refractory period. Changes in refractoriness (determined by either
altered recovery from inactivation or altered action potential duration) can be
important in the genesis or sup-pression of certain arrhythmias. Another
important effect of less negative resting potential is prolongation of this
recovery time, as shown in Figure 14ÔÇô4 (right panel). The prolongation of
recovery time is reflected in an increase in the effective refractory period.
A
brief, sudden, depolarizing stimulus, whether caused by a propagating action
potential or by an external electrode arrangement, causes the opening of large
numbers of activation gates before a significant number of inactivation gates
can close. In contrast, slow reduction (depolarization) of the resting
poten-tial, whether brought about by hyperkalemia, sodium pump blockade, or
ischemic cell damage, results in depressed sodium currents during the upstrokes
of action potentials. Depolarization of the resting potential to levels
positive to 55 mV abolishes sodium currents, since all sodium channels are
inactivated. However, such severely depolarized cells have been found to
sup-port special action potentials under circumstances that increase calcium
permeability or decrease potassium permeability. These ÔÇ£slow responsesÔÇØÔÇöslow
upstroke velocity and slow conductionÔÇö depend on a calcium inward current and
constitute the normal electrical activity in the SA and AV nodes, since these
tissues have a normal resting potential in the range of 50 to 70 mV. Slow
responses may also be important for certain arrhythmias.
Modern
techniques of molecular biology and electrophysiol-ogy can identify multiple
subtypes of calcium and potassium channels. One way in which such subtypes may
differ is in sensi-tivity to drug effects, so drugs targeting specific channel
subtypes may be developed in the future.
Related Topics