Chapter: Basic & Clinical Pharmacology : Agents Used in Cardiac Arrhythmias
Basic Pharmacology of the Antiarrhythmic Agents
BASIC
PHARMACOLOGY OF THE ANTIARRHYTHMIC AGENTS
Mechanisms of Action
Arrhythmias
are caused by abnormal pacemaker activity or abnormal impulse propagation.
Thus, the aim of therapy of the arrhyth-mias is to reduce ectopic pacemaker
activity and modify conduction or refractoriness in reentry circuits to disable
circus movement. The major pharmacologic mechanisms currently available for
accomplishing these goals are (1) sodium channel blockade, (2) blockade of
sympathetic autonomic effects in the heart, (3) pro-longation of the effective
refractory period, and (4) calcium chan-nel blockade.
Antiarrhythmic
drugs decrease the automaticity of ectopic pace-makers more than that of the SA
node. They also reduce conduction and excitability and increase the refractory
period to a greater extent in depolarized tissue than in normally polarized
tissue. This is accomplished chiefly by selectively blocking the sodium or
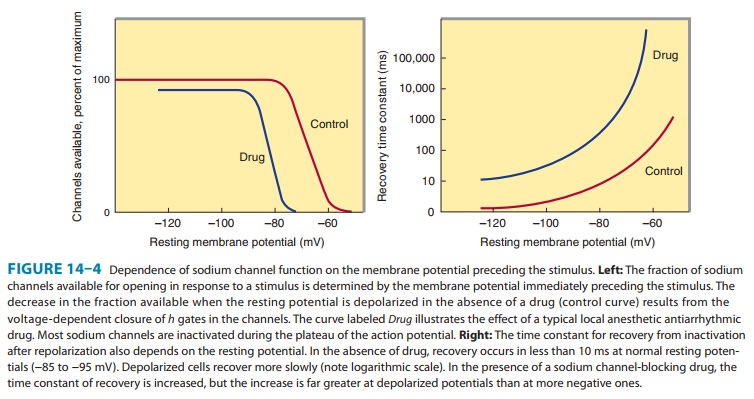
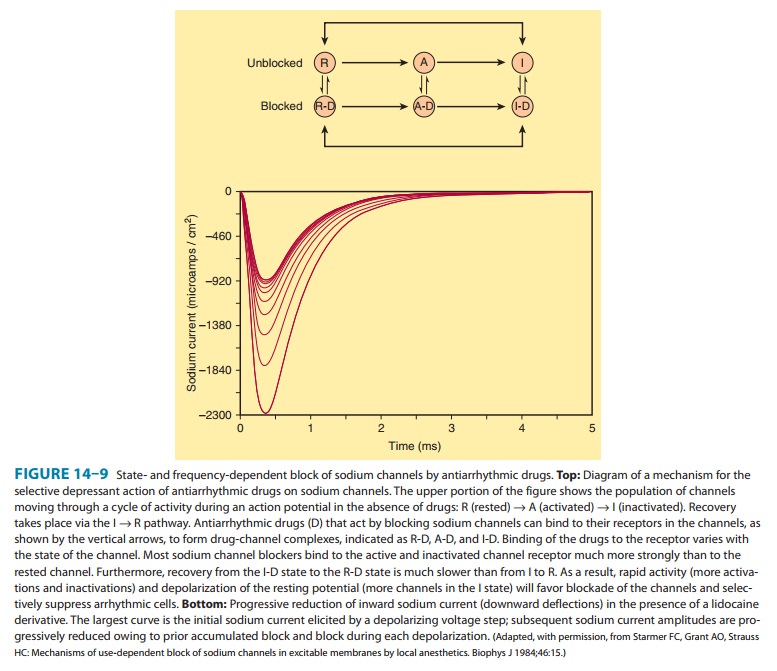
calcium channels of depolarized cells (Figure 14ÔÇô9). Therapeutically useful channel-blocking
drugs bind readily to activated channels (ie, dur-ing phase 0) or inactivated
channels (ie, during phase 2) but bind poorly or not at all to rested channels.
Therefore, these drugs block electrical activity when there is a fast
tachycardia (many channel activations and inactivations per unit time) or when
there is signifi-cant loss of resting potential (many inactivated channels
during rest). This type of drug action is often described as use-dependent or state-dependent; that is, channels that are being used frequently,
or in an inactivated state, are more susceptible to block. Channels in normal
cells that become blocked by a drug during normal activation-inactivation
cycles will rapidly lose the drug from the receptors during the resting portion
of the cycle (Figure 14ÔÇô9). Channels in myocardium that is chronically
depolarized (ie, has a resting potential more positive than 75 mV) recover
from block very slowly if at all (see also right panel, Figure 14ÔÇô4).
In
cells with abnormal automaticity, most of these drugs reduce the phase 4 slope
by blocking either sodium or calcium channels, thereby reducing the ratio of
sodium (or calcium) permeability to potassium permeability. As a result, the
membrane potential during phase 4 stabilizes closer to the potassium
equilibrium potential. In addition, some agents may increase the threshold
(make it more positive). ╬▓-AdrenoceptorÔÇôblocking drugs indirectly reduce
the phase 4 slope by blocking the positive chronotropic action of
nor-epinephrine in the heart.
In
reentry arrhythmias, which depend on critically depressed conduction, most
antiarrhythmic agents slow conduction fur-ther by one or both of two
mechanisms: (1) steady-state reduction in the number of available unblocked
channels, which reduces the excitatory currents to a level below that required
for propagation (Figure 14ÔÇô4, left); and (2) prolonga-tion of recovery time of
the channels still able to reach the rested and available state, which
increases the effective refractory period (Figure 14ÔÇô4, right). As a result,
early extrasystoles are unable to propagate at all; later impulses propagate
more slowly and are subject to bidirectional conduction block.
By
these mechanisms, antiarrhythmic drugs can suppress ectopic automaticity and
abnormal conduction occurring in depolarized cellsÔÇörendering them electrically
silentÔÇöwhile minimally affecting the electrical activity in normally polarized
parts of the heart. However, as dosage is increased, these agents also depress
conduction in normal tissue, eventually resulting in drug-in-duced arrhythmias. Furthermore, a drug concentration that
istherapeutic (antiarrhythmic) under the initial circumstances of treatment may
become ÔÇ£proarrhythmicÔÇØ (arrhythmogenic) during fast heart rates (more
development of block), acidosis (slower recovery from block for most drugs),
hyperkalemia, or ischemia.
Related Topics