Chapter: Clinical Anesthesiology: Perioperative & Critical Care Medicine: Management of Patients with Fluid & Electrolyte Disturbances
Hyperkalemia
HYPERKALEMIA
Hyperkalemia exists when plasma [K+] exceeds 5.5 mEq/L.
Hyperkalemia rarely occurs in nor-mal individuals because of the kidney’s
capability to excrete large potassium loads. When potassium intake is increased
slowly, the kidneys can excrete as much as 500 mEq of K+ per day. The
sympathetic nervous system and insulin secretion also play important roles in
preventing acute increases in plasma [K+] following acquired potassium loads.
Hyperkalemia can result from (1) an
intercom-partmental shift of potassium ions, (2) decreased urinary excretion of
potassium, or, rarely, (3) an increased potassium intake (Table
49–10). Mea-surements of plasma potassium
concentration can be spuriously elevated if red cells hemolyze in a blood
specimen. In vitro release of potassium from white cells in a blood specimen
can also falsely indi-cate increased levels in the measured plasma [K+] when the leukocyte
count exceeds 70,000 × 109/L.
A similar release of potassium from platelets occurs when the platelet count
exceeds 1,000,000 × 109/L.
Hyperkalemia due to Extracellular Movement of Potassium
Movement of K+ out of cells can be seen with aci-dosis,
cell lysis following chemotherapy, hemolysis,rhabdomyolysis, massive tissue
trauma, hyperosmo-lality, digitalis overdoses, during episodes of hyper-kalemic
periodic paralysis, and with administration of succinylcholine, β2-adrenergic
blockers, and argi-nine hydrochloride. The average increase in plasma [K+] of 0.5 mEq/L following
succinylcholine admin-istration can be exaggerated in patients with large burns
or severe muscle trauma and in those with muscle denervation, and its use in
these settings should be avoided.
β2-Adrenergic blockade
accentuates the increasein plasma [K+] that occurs following exercise.
Digoxin inhibits Na+–K+-ATPase in cell membranes, and
digoxin overdose has been reported to cause hyperkalemia in some patients.
Arginine hydro-chloride, which is used to treat metabolic alkalosis, evaluate
pituitary growth hormone reserve, and as a performance-enhancing supplement by
athletes, can cause hyperkalemia as the cationic arginine ions enter cells and
potassium ions move out to maintain electroneutrality.
Hyperkalemia due to Decreased Renal Excretion of Potassium
Decreased renal excretion of potassium can result from (1) marked
reductions in glomerular filtration,decreased aldosterone activity, or (3) a
defect in potassium secretion in the distal nephron.
Glomerular filtration rates less than 5 mL/min are nearly always
associated with hyperkalemia. Patients with lesser degrees of renal impairment
can also read-ily develop hyperkalemia when faced with increased potassium
loads (dietary, catabolic, or iatrogenic). Uremia may also impair Na+–K+-ATPase activity.
Hyperkalemia due to decreased aldosterone
activity can result from a primary defect in adre-nal hormone synthesis or a
defect in the renin– aldosterone system. Patients with primary adrenal
insufficiency (Addison’s disease) and those with isolated 21-hydroxylase
adrenal enzyme deficiency have marked impairment of aldosterone synthesis.
Patients with the syndrome of isolated hypoaldoste-ronism (also called
hyporeninemic hypoaldosteron-ism, or type IV renal tubular acidosis) are usually
diabetics with some degree of renal impairment; they have an impaired ability
to increase aldosterone secretion in response to hyperkalemia. Although usually
asymptomatic, these patients develop hyper-kalemia when they increase their
potassium intake or when given potassium-sparing diuretics. They also often
have varying degrees of Na+ wasting and a hyperchloremic metabolic acidosis. Similar findings have
been reported in patients with AIDS who have relative adrenal insufficiency due
to cytomegalovi-rus infection.
Drugs interfering with the renin–aldosterone system have the potential
to cause hyperkalemia, particularly in the presence of any degree of renal
impairment. Nonsteroidal antiinflammatory drugs (NSAIDs) inhibit
prostaglandin-mediated renin release. Angiotensin-converting enzyme (ACE)
inhibitors interfere with angiotensin II–mediated release of aldosterone. Large
doses of heparin can interfere with aldosterone secretion. The
potassium-sparing diuretic spironolactone directly antagonizes aldosterone activity
at the kidneys.
Decreased renal excretion of potassium can also occur as a result of an
intrinsic or acquired defect in the distal nephron’s ability to secrete
potassium. Such defects may occur even in the presence of normal renal function
and are char-acteristically unresponsive to mineralocorticoid therapy. The
kidneys of patients with pseudohy-poaldosteronism display an intrinsic
resistance to aldosterone. Acquired defects have been associ-ated with systemic
lupus erythematosus, sickle cell anemia, obstructive uropathies, and
cyclosporine nephropathy in transplanted kidneys.
Hyperkalemia due to Increased Potassium Intake
Increased potassium loads rarely cause hyperka-lemia in normal individuals unless large amounts are given rapidly and intravenously. Hyperkale-mia, however, may be seen when potassium intake is increased in patients receiving β blockers or in patients with renal impairment. Unrecognized sources of potassium include potassium penicillin, sodium substitutes (primarily potassium salts), and transfusion of stored whole blood. The plasma [K+] in a unit of whole blood can increase to 30 mEq/L after 21 days of storage. The risk of hyperkalemia from multiple transfusions is reduced, although not eliminated, by minimizing the volume of plasma given through the use of packed red blood cell trans-fusions .
Clinical Manifestations of Hyperkalemia
The most important effects of hyperkalemia
are on skeletal and cardiac muscle. Skeletal muscle weak-ness is generally not
seen until plasma [K+] is greater than 8 mEq/L, and is due to sustained spontaneous
depolarization and inactivation of Na+ channels of muscle membrane, eventually
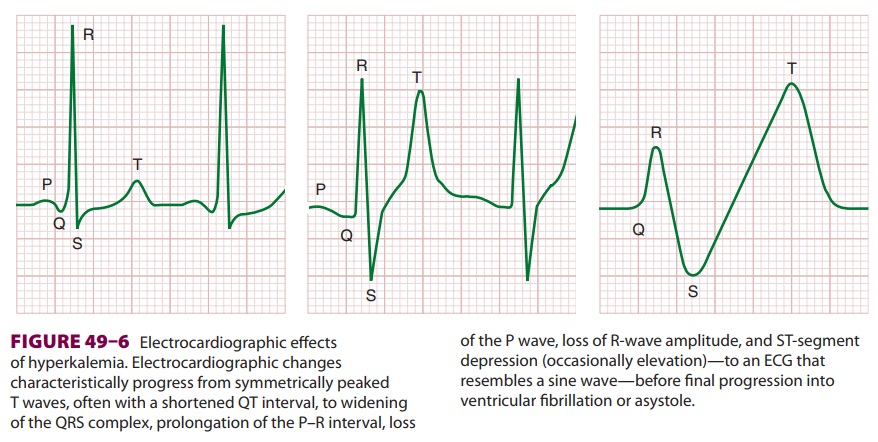
resulting in paralysis. Cardiac manifestations ( Figure 49–6) are primarily due to delayed depolarization, and are consistently
present when plasma [K+] is greater than 7 mEq/L. ECG changes characteristically progress
sequen-tially from symmetrically peaked T waves (often with a shortened QT
interval) → widening of the QRS
complex → prolongation of the P–R
interval → loss of the P wave → loss of R-wave amplitude → ST-segment depression (occasionally
elevation) → an ECG that resembles a
sine wave, before progres-sion to ventricular fibrillation and asystole.
Contrac-tility may be relatively well preserved until late in the course of
progressive hyperkalemia. Hypocalcemia, hyponatremia, and acidosis accentuate
the cardiac effects of hyperkalemia.
Treatment of Hyperkalemia
Because of its lethal potential, hyperkalemia
exceeding 6 mEq/L should always be corrected. Treatment is directed to reversal
of cardiac manifestations and skeletal muscle weakness, and to restoration of
normal plasma [K +]. Therapeu-tic modalities employed depend on the cause of hyperkalemia
and the severity of manifestations. Hyperkalemia associated with
hypoaldosteronism can be treated with mineralocorticoid replace-ment. Drugs
contributing to hyperkalemia should be discontinued and sources of increased
potas-sium intake reduced or stopped.
Calcium (5–10 mL of 10% calcium gluconate or
3–5 mL of 10% calcium chloride) partially antago-nizes the cardiac effects of
hyperkalemia and is use-ful in patients with marked hyperkalemia. Its effects
are rapid but short lived. Care must be exercised in administering calcium to
patients taking digoxin, as calcium potentiates digoxin toxicity.
When metabolic acidosis is present, intrave-nous sodium bicarbonate
(usually 45 mEq) will pro-mote cellular uptake of potassium and can decrease
plasma [K+] within
15 min. β Agonists promote cellular
uptake of potassium and may be useful in acute hyperkalemia associated with
massive transfu-sions; low-dose epinephrine infusion often rapidlydecreases
plasma [K+] and
provides inotropic sup-port in this setting. An intravenous infusion of
glu-cose and insulin (30–50 g of glucose with 10 units of insulin) is also
effective in promoting cellular uptake of potassium and lowering plasma [K+], but may take up to 1 h for
peak effect.
For patients with some renal function,
furose-mide is a useful adjunct in increasing urinary excre-tion of potassium.
In the absence of renal function, elimination of excess potassium can be
accom-plished only with nonabsorbable cation-exchange resins such as oral or
rectal sodium polystyrene sul-fonate (Kayexalate). Each gram of resin binds up
to 1 mEq of K + and releases 1.5 mEq of Na +; the oral dose is 20 g in 100 mL of 20%
sorbitol.
Dialysis is indicated in symptomatic patients
with severe or refractory hyperkalemia. Hemodialy-sis is faster and more
effective than peritoneal dialy-sis in decreasing plasma [K+]. Maximal potassium
removal with hemodialysis approaches 50 mEq/h, compared with 10–15 mEq/h for
peritoneal dialysis.
Anesthetic Considerations
Elective surgery should not be undertaken in
patients with significant hyperkalemia.
Anesthetic management of hyperkalemic surgical patients is directed at
both lowering the plasma potassium con-centration and preventing any further
increases. The ECG should be carefully monitored. Succinylcholine is
contraindicated, as is the use of any potassium-containing intravenous
solutions such as lactated Ringer’s injection. The avoidance of metabolic or
respiratory acidosis is critical to prevent further increases in plasma [K+]. Ventilation should be con-trolled under general anesthesia, and mild
hyper-ventilation may be desirable. Lastly, neuromuscular function should be
monitored closely, as hyperkale-mia can accentuate the effects of NMBs.
Related Topics