Chapter: Biology of Disease: Disorders of Water, Electrolytes and Urate Balances
Disorders of Urate Metabolism
DISORDERS OF URATE METABOLISM
In humans, the end product of the metabolism of the purines,
adenine and guanine is urate (Figure 8.19).
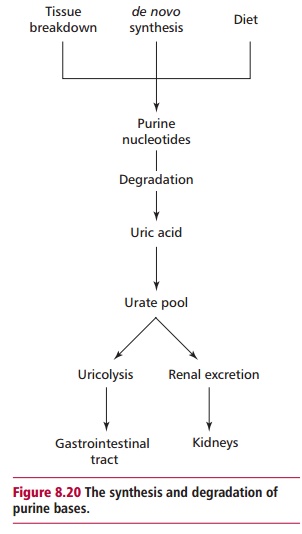
There are three sources of purines namely diet, the breakdown of endogenous
nucleotides and nucleic acids and de novo
synthesis. Most dietary nucleic acids are ingested in the form
ofnucleoproteins from which urate is produced by the GIT . The degradation and de novo synthesis of purines are linked
(Figure 8.20). The body urate pool,
and therefore plasma concentration, depends upon the relative rates of urate
formation and excretion. Both the kidneys and the GIT excrete urate with renal
excretion accounting for approximately 66% of the total. Almost all the urate
is filtered at the glomerulus but most is reabsorbed by the proximal tubule.
However, both reabsorption and secretion occur in the distal tubule, so that
the net effect is to excrete about 10% of the urate. Urate secreted into the
GIT is metabolized to CO2 and NH3 by bacterial action or uricolysis.

The reference range for serum urate is 0.1 to 0.4 mmol dm–3.
However, there is a wide variation in the concentration of urate in plasma or
serum even in health. Plasma urate concentration tends to be higher in males
than females, is highest in obese individuals, those from affluent social
classes and those with a high protein and alcohol intake. Thus hyperuricemia is
defined as a concentration greater than 0.42 mmol dm–3 in men and
more than 0.36 mmol dm–3 in women.
Hyperuricemia may arise as a result of increased production of
uric acid or decreased excretion or both. Excessive synthesis may occur because
of a defective synthetic metabolic pathway, stimulation of de novo purine synthesis by alcohol or by increased nucleic acid
turnover, as in malignant disease, or the use of cytotoxic drugs. An excessive
dietary intake of purines will also produce hyperuricemia. A decreased urate
excretion may be due to a reduced GFR giving rise to hyperuricemia. Increased
proximal tubular reabsorption and decreased distal tubular secretion of urate
have similar effects. Lactate and A-hydroxybutyrate compete with urate for excretion by the distal
tubule. Therefore lactic acidosis or ketosis are often associated with
hyperuricemia. Some drugs, for example lowdoses of aspirin, can inhibit the
distal tubular secretion of urate causing hyperuricemia.
Urate has low solubility and the ECF easily becomes saturated at
concentrations just above the upper limit of the reference range. There is a
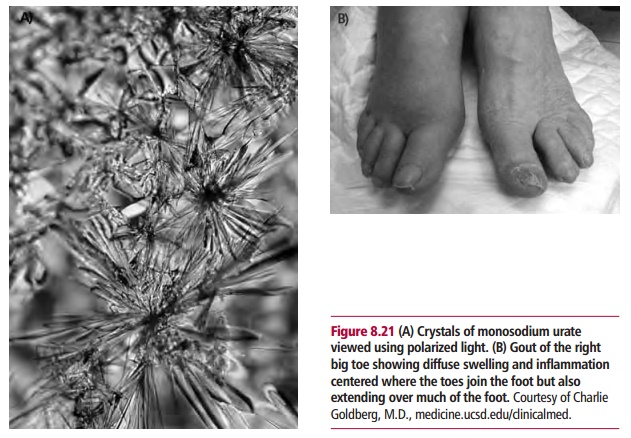
tendency for crystalline monosodium urate to form in people with hyperuricemia,
giving rise to gout. Crystals of monosodium urate (Figure8.21(A)) tend to form in cartilage and synovial fluid of
joints and particularlythose of the big toe causing gout (Figure 8.21(B)). The crystals are phagocytosed by neutrophil
leukocytes and may cause damage to lysosomal membranes within these cells . As
a consequence, lysosomal contents are released, causing damage to both
leukocytes and surrounding tissues with an associated inflammatory response .
Gout may be primary, with no known cause, or secondary, as a consequence of
another disorder. Primary gout is characterized by recurrent attacks of
arthritis. It is more common in men than women. The metabolic defect in
patients is unknown but a number of abnormalities may be responsible for the
overproduction of urate and therefore increased urinary urate output. In many
patients there is a combined defect of urate overproduction together with its
impaired renal excretion. Patients with primary gout often have deposits of
urate in their soft tissues and some can develop renal stones composed of urate
salts. The risk that a normal person will develop gout varies with their urate
concentration. The annual incidence of gout in men is low, about 0.1%, when the
urate concentration is less than 0.42 mmol dm–3. This increases to
0.6% when the concentration is 0.42–0.54 mmol dm–3 and 5% when urate
concentrations are greater than 0.54 mmol dm–3. The reason for the
onset of acute attacks in gout is unclear since a sharp rise in the
concentration of urate is not usually demonstrable.
Secondary gout is rare but can arise from a number of other
disorders including myeloproliferative disorders such as polycythemia vera,
where the hyperuricemia is due to an increased cell turnover, the use of
cytotoxic drug therapy that increases cell destruction and the breakdown of
nucleic acids, and psoriasis with its increased turnover of skin cells.
A diagnosis of gout is made on clinical grounds, a demonstration
of hyper-uricemia and a satisfactory response to uricosuric drugs. A high
plasma urate concentration does not always mean that the patient has gout, that
is high plasma urate concentration makes the diagnosis of gout more likely,
whereas a consistently low plasma urate concentration excludes the diagnosis.
To confirm diagnosis it is necessary to aspirate the joint fluid during an
acute attack. The finding of urate crystals, 2–10 µm long and needle shaped,
within neutrophils will confirm the diagnosis.
Anti-inflammatory drugs, such as indomethacin, are used to treat
acute attacks of gout but have no effect on the hyperuricemia which is treated
with a diet low in protein and alcohol. Urate lowering drugs, for example
allopurinol, that prevent the formation of urate and decrease de novo synthesis of purines, are used
in long-term treatment or when plasma urate levels are persistently higher than
0.6 mmol dm–3.
Hypouricemia, where the concentration of urate in serum is below
the reference range, is uncommon and not of clinical significance. Its
occurrence is due to a decreased urate synthesis, as in congenital xanthine
oxidase deficiency and severe liver disease, or to increased excretion of urate
as seen in renal tubular disorders, such as the Fanconi syndrome. Hypouricemia
may also result from excessive use of drugs such as allopurinol.
Related Topics