Chapter: Biology of Disease: Disorders of Water, Electrolytes and Urate Balances
Disorders of Phosphate Homeostasis
DISORDERS OF PHOSPHATE
HOMEOSTASIS
Phosphate (Pi) combines with Ca2+ to form
hydroxyapatite, the mineral component of bone and teeth and is also required
for some enzymic activities, oxidative phosphorylation and the synthesis of
2,3-bisphosphoglycerate that regulates the dissociation of oxyhemoglobin , the
excretion of H+ and for cell membrane integrity. The daily intake of
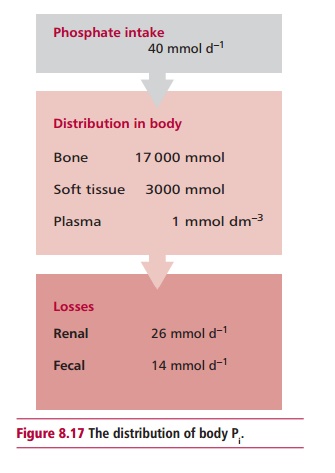
Pi is about 40 mmol. The kidneys lose approximately 26 mmol daily
and 14 mmol are lost in feces. The total body content of Pi in the
average male is over 20 000 mmol (Figure
8.17) with 17 000 occurring in bone and 3000 in soft tissues, largely
attached to lipids and proteins. Thus about 85% occurs in bone while the ICF
and the ECF contain 15% and 0.1% respectively. The plasma concentration is
about 1 mmol dm–3. Approximately 80% of the plasma content occurs as
free inorganic Pi, 15% is protein-bound and about 5% is complexed
with Ca2+ and Mg2+. Parathyroid hormone (Figure 8.11) and the hormone, calcitriol,
control the homeostasis of Pi; the former decreasing the
reabsorption by the kidneys and reducing its plasma concentration, the latter
stimulating Pi absorption in the GIT and increasing the
concentration.
The reference range for total serum Pi is 0.8–1.4
mmol dm–3 but a higher reference range applies in infancy and
childhood. Hyperphosphatemia and hypophosphatemia are used to describe
concentrations above and below the reference range respectively.
Hypophosphatemia causes more damage than hyperphosphatemia but, fortunately, is
less common.
Hyperphosphatemia may cause metastatic calcification, for
example the deposition of calcium phosphate in soft tissues as the excess Pi
precipitates with Ca2+ and causes hypocalcemia and tetany in
affected patients. The commonest cause of hyperphosphatemia is renal failure
where the GFR and Pi excretion decline. Hypoparathyroidism reduces
renal excretion of Pi giving rise to hyperphosphatemia. In diabetic
ketoacidosis , a deficiency of insulin prevents the uptake of Pi by
cells leading to hyperphosphatemia. Other causes are an increased intake of Pi
or its release from damaged cells in intravascular hemolysis. Indeed, any
condition where there is increased turnover of cells, for example following
treatment of malignant disease with chemotherapy, results in release of Pi
during cell destruction. Excessive intake, either oral or intravenous, is a
rare cause and is more likely when there is also renal failure as in pseudohypoparathyroidism
where there is resistance by the kidneys to PTH that decreases their excretion
of Pi. A delay in the separation of plasma or serum from blood
before analysis for Pi or hemolysis of a blood sample prior to its
analysis can indicate artefactual hyperphosphatemia but this does not reflect
the true clinical situation.
A number of biochemical tests are useful when investigating
hyperphosphatemia. These include determining the concentrations of Pi,
Ca2+, urea and creatinine in serum and the concentration of Pi
in urine. The following strategy has proved useful in investigating obscure
causes of hyperphosphatemia. First, it is necessary to exclude artefactual
causes. Secondly, serum concentrations of creatinine and urea should be
determined to exclude renal failure. If the serum concentration of Ca2+
is normal or above reference values, vitamin D intoxication or untreated
diabetes mellitus should be considered. Thirdly, if the plasma or serum
concentration of Ca2+ is low, then hypoparathyroidism should be
investigated. Finally, if the urinary concentration of Pi is low,
then hypoparathyroidism is, again, a consideration, whereas a high urinary
concentration indicates increased intake, malignancy or intravascular
hemolysis. Patients with hyperphosphatemia are managed by treating the
underlying cause wherever possible. The oral intake of aluminum, Ca2+
and Mg2+ salts may be used as these can bind Pi in the
GIT reducing its absorption.
The clinical features of hypophosphatemia include paresthesiae,
ataxia, coma, osteomalacia and muscle weakness. There may be increased
susceptibility to infection possibly due to defective phagocytosis. The causes
of hypophosphatemia are varied. Vitamin D deficiency results in a decreased
synthesis of calcitriol and therefore decreased Pi absorption in the
GIT. Increased renal loss of Pi may occur in primary
hyperparathyroidism where increased secretion of PTH causes excessive renal
loss of Pi. Certain diuretics that increase renal loss of Pi
can cause hypophosphatemia. It may also occur during the recovery phase of
diabetic ketoacidosis when patients are administered insulin, which promotes
cellular uptake of Pi. Total body Pi may be depleted as a
consequence of osmotic diuresis. There are a number of rare causes of
hypophosphatemia. These include an inadequate dietary intake usually associated
with parenteral nutrition, or when agents, such as aluminum hydroxide are used
as antacids and prevent its absorption in the GIT, and in chronic alcoholics
who have a complex and multifactorial condition with poor diet and reduced GIT
absorption .
Determination of the serum concentrations of Pi and Ca2+ and the urinary concentration of Pi are useful in investigating hypophosphatemia. The following strategy may be used when its cause is not obvious.
First, exclude causes such as alkalosis and chronic alcoholism. Secondly, a
reduced urinary Pi suggests decreased dietary or parenteral intakes
or increased cellular uptake, for example in insulin therapy. Thirdly, if the
urinary concentration of Pi is above its reference range then
excessive renal losses are occurring and the concentration of Ca2+
in the plasma or serum should be determined. If this is increased, then primary
hyperparathyroidism or malignancy may be present. If, however, the
concentration is low or normal, renal defects or inappropriate diuretic therapy
are considerations. Hypophosphatemia should be managed by treating the
underlying cause wherever possible. In some situations it may be necessary to
administer oral or parenteral Pi.
Related Topics