Chapter: Clinical Dermatology: Connective tissue disorders
Dermatomyositis
Dermatomyositis
Dermatomyositis
is a subset of polymyositis with distinctive skin changes. There are adult and
juvenile types (Table 10.2). The cause is unknown but an auto-immune mechanism
seems likely. Autoantibodies to striated muscle are found. When starting after
the age of 40, dermatomyositis may signal an internal malig-nancy. Presumably,
the epitopes of some tumour anti-gens are so similar to those of muscle
antigens that antibodies directed against the tumour cross-react with muscle
cells and initiate the disease in a few adults with internal malignancy.
Serological evidence for acute toxoplasmosis in polymyositis-dermato-myositis
was found in one series.

Presentation
The
skin signs are characteristic. Typical patients have a faint lilac
discoloration around their eyes (sometimes called ‘heliotrope’ because of the
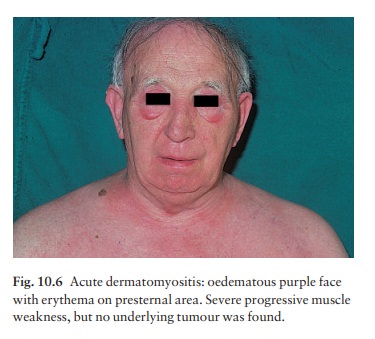
colour of the flower). This is associated with malar erythema and oedema (Fig.
10.6) and, sometimes, less striking erythema of the neck and presternal area.
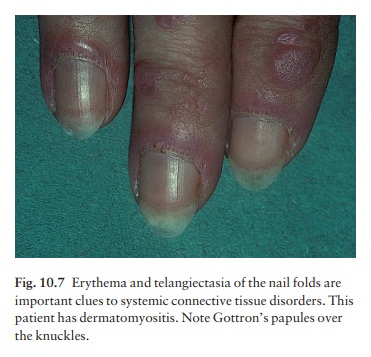
Most patients also develop lilac slightly atrophic papules over the knuckles of
their fingers (Gottron’s papules), streaks of erythema over the extensor
tendons of the hand, peri-ungual telangiectasia and ragged cuticles (Fig.
10.7). The skin signs usually appear at the same time as the muscle symptoms
but, occasionally, appear months or even years earlier. Sometimes, the skin
signs appear in isolation. Many, but not all, patients have weakness of
proximal muscles. Climbing stairs, getting up from chairs and combing the hair
become difficult.
Course
In children the disorder is often self-limiting, but in adults it may be prolonged and progressive. Raynaud’s phenomenon, arthralgia, dysphagia and calcinosis may follow. The rash may become scaly and, rarely, itchy; eventually that on the light-exposed areas and overly-ing involved muscles develops poikiloderma. Features of mixed connective disease may develop. The presence of calcinosis suggests a good prognosis.
Complications
Myositis
may lead to permanent weakness and immo-bility, and inflammation to
contractures or cutaneous calcinosis. Some die from progessive and severe
myopathy.
Differential diagnosis
Other
connective tissue disorders may look similar, particularly mixed connective
tissue disease and SLE. In LE, the
finger lesions favour the skin between the knuckles whereas in dermatomyositis
the knuckles are preferred. Toxoplasmosis may cause a dermatomyositis-like
syndrome. Myopathy can be a side-effect of systemic steroids, so weakness is
not always caused by the disease itself.
Investigations
About
30% of adults with dermatomyositis also have an underlying malignancy. Their
dermatomy-ositis coincides with the onset of the tumour and may improve if it
is removed. Adult dermatomyositis or polymyositis therefore requires a search
for such an underlying malignancy. The levels of muscle enzymes such as
aldolase and creatinine phosphokinase (CPK) are often elevated.
Electromyography (EMG) detects muscle abnormalities, and biopsy of an affected
muscle shows inflammation and destruction. Sur-prisingly, the ESR is often
normal and antinuclear antibodies may not be detected. Toxoplasmosis should be
excluded by serology.
Treatment
Systemic
steroids, often in high doses (e.g. prednisolone mg/day for an average adult;),
are the cornerstone of treatment and protect the muscles from destruction. A
maintenance regimen may be needed for several years. Immunosuppressive agents,
such as azathioprine, also help to control the condition and to reduce the high
steroid dose. Cyclosporin and
methotrexate have proved useful
alternatives in stubborn cases. Maintenance treatment is adjusted according to
clinical response and CPK level. As in SLE, intravenous gamma globulin
infusions seem promising. Long-term and regular follow-up is necessary.
Related Topics