Chapter: Biology of Disease: Genetic Diseases
Consequences of an Enzyme Deficiency
CONSEQUENCES OF AN ENZYME DEFICIENCY
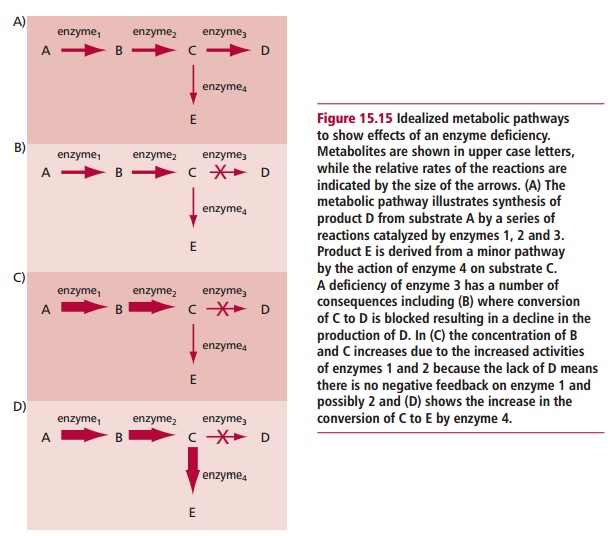
In inherited metabolic disorders caused by a complete or partial deficiency of an enzyme that controls a particular reaction in a metabolic pathway, the clinical features are a consequence of the enzyme deficiency. Figure 15.15 shows how this may occur. Symptoms arise from a lack of product D if this isan essential substance and alternative pathways for its synthesis do not exist. Moreover, an accumulation of precursor C will occur if the enzyme is absent and may produce clinical features if the substrate is toxic when it accumulates. Intermediates of the pathway may also accumulate especially since there will be no negative feedback effect as the final product of the pathway is absent and cannot inhibit the first enzyme of the pathway. Finally, accumulation of a product of a minor pathway, E, may occur and if this is toxic in excess it may produce clinical features.
The treatment of inherited metabolic disorders aims at trying to prevent the accumulation of precursor(s) and provide the necessary product of the pathway. The removal of toxic products of any minor pathways may also be necessary. Future strategies are aimed at replacing the deficient enzyme or correcting the defective gene by gene therapy. Many inherited metabolic disorders caused by a deficiency of an enzyme are known (Table 15.4); one of the most thoroughly documented is phenylketonuria.
Phenylketonuria
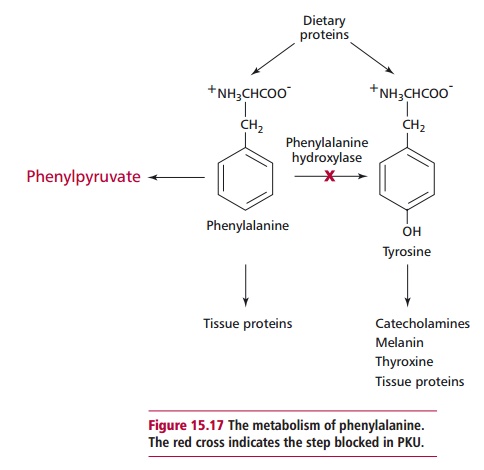
Phenylketonuria (PKU) is the commonest disorder of amino acid metabolism. It has an autosomal recessive mode of inheritance, which leads to a deficiency, mostly in the liver, of phenylalanine hydroxylase (Figure 15.16), which catalyzes the hydroxylation of phenylalanine to tyrosine (Figure 15.17). Tyrosine is required for the synthesis of proteins, the pigment melanin, thyroxine and the catecholamine hormones . However, if the enzyme is absent, then phenylalanine and its metabolites accumulate and are toxic to the developing brain. The manner in which damage occurs is not completely understood but it is believed that hyperphenylalaninemia interferes with brain amino acid metabolism and inhibits the release of neurotransmitters. There is also an increase in the level of phenylpyruvic acid, a phenylketone, which is normally a minor metabolite of phenylalanine (Figure 15.17). Excess phenylpyruvic acid is excreted in urine, hence the name phenylketonuria. The incidence of PKU is one in 10 000 in the UK; in other countries it varies from one in 5000 to one in 20 000 births.
The clinical features of PKU are absent at birth but develop within a few days if the newborn is untreated. These signs include a characteristic mousy odor, irritability, poor feeding, vomiting, eczema, mental retardation, as well as a pale skin, fair hair and blue eyes due to decreased melanin synthesis. The most serious of these features is irreversible mental retardation which develops within three to six months following birth. A diagnosis of PKU is made on the
demonstration of a concentration of phenylalanine in the serum in excess of 0.7 mmol dm–3, compared with the reference range value of less than 0.1 mmol dm–3. Clinical determinations are performed a few days following birth since it is vital to begin any treatments as soon as possible. The management of PKU involves restricting the dietary intake of phenylalanine so that the serum phenylalanine concentration does not exceed the limits shown in Table 15.5.
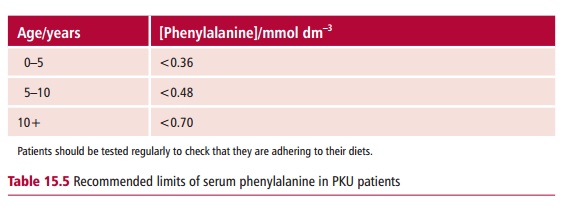
In the early stages of life, when the brain is developing rapidly, strict control of phenylalanine concentrations must be imposed to prevent brain damage. Commercially prepared diets with a low phenylalanine content are available. The concentration of this amino acid is low but cannot be zero since phenylalanine is an essential amino acid and some must be provided in the diet to support protein synthesis. Tyrosine is not an essential amino acid unless phenylalanine intake is limited. Therefore adequate quantities of tyrosine must also be provided in the diet of PKU patients. Regular monitoring of treatment is advisable. Blood is collected as a dried spot or as liquid plasma for laboratory analysis. The diet can become somewhat less rigorous after the age of 10, although many clinicians now believe that dietary restriction should be continued throughout life.
Children who have been diagnosed shortly after birth and properly treated by dietary management develop normally. Early treatment is crucial because the IQ of an affected individual rarely exceeds 70 and brain damage caused by untreated PKU is irreversible. Strict dietary control is also necessary in pregnant women who have PKU, since maternal hyperphenylalaninemia can affect the fetus in utero, even if the fetus itself does not have PKU. Mental retardation and congenital abnormalities can occur in a large proportion of these infants.
Related Topics