Chapter: Biology of Disease: Genetic Diseases
Chromosomal Mutations or Aberrations
CHROMOSOMAL MUTATIONS OR ABERRATIONS
Chromosomal mutations include structural changes within a single chromosome or changes in the number of chromosomes present. Structural mutations occur when chromosomes break and, although in general repair mechanisms rejoin the two ends to restore rapidly the original structure, if more than one break occurs, the repair mechanisms are unable to distinguish between the broken ends and portions of different chromosomes may be joined together. This can lead to one of four major types of chromosomal structural aberration or mutation. In deletions, a section of the DNA is lost; in duplications, one or more extra copies of a segment of DNA occur in a chromosome; in inversions, there is a reversal of direction of a portion of the DNA in the chromosome and in translocations, segments of the DNA are moved to a different chromosome. Deletions and duplications change the amount of DNA in a chromosome. Inversions and translocations change the arrangement of bases in a length of DNA but do not change the amount of DNA present in the chromosome.
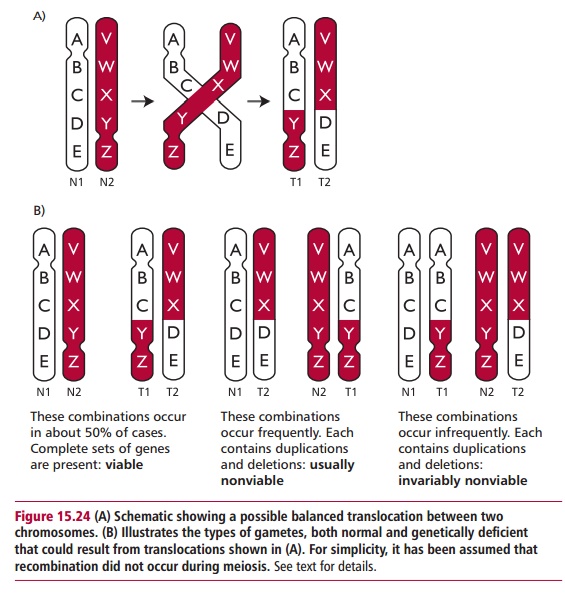
If the exchange of chromosomal material during a translocation and inversion does not involve breaks within a gene or alter the amount of DNA, then the individual will be clinically normal and is said to have a balanced translocation. However, if the structural alteration occurs in the gonads, even a balanced translocation has clinical significance for future generations since it may lead to offspring who are chromosomally unbalanced, that is, who have lost DNA (Figure 15.24). Since most fetuses with unbalanced translocations tend to be spontaneously aborted, people with balanced translocations may only attract clinical attention if they and their partner are investigated because of a history of miscarriages. Infants with unbalanced translocations that do survive are mentally retarded and show multiple dysmorphic features, that is alterations (abnormalities) to the accepted appearance. Specific disorders can also occur when discrete genes are damaged at the translocation fractures, the resulting disorder being dependent on which genes are damaged.
DELETIONS
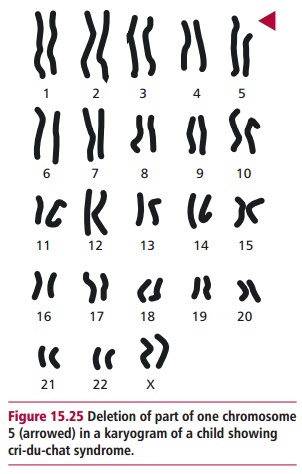
Deletion of part of a chromosome can arise between two breakpoints as a result of unequal crossing over during meiosis or as a result of a parental translocation. Several clinical disorders are caused by deletions. In many cases, the abnormalities only occur in individuals who are heterozygous since the homozygous condition is lethal, especially if the deleted portion of the chromosome is large. However, any chromosomal deletion that canbe observed microscopically almost invariably produces a phenotype with multiple abnormal features and mental retardation because of the absence of expression of the deleted genes. For example, cri-du-chat syndrome is a heterozygous condition that occurs in about one in 50 000 births. Infants with this syndrome show anatomical malformations including gastrointestinal and cardiac complications and are often mentally retarded. The glottis and larynx also develop abnormally giving the characteristic cry, similar to the meowing of a cat, which names the syndrome. Cri-du-chat syndrome is caused by a deletion of part of the short arm of chromosome 5 (46,5p-, Figure 15.25). The length of the deleted portion varies; the longer the deletion the more defective the syndrome in surviving children. The effects of the syndrome are severe, although individuals who receive good home care and early special schooling can walk and communicate verbally and develop self-care skills.
Prader-Willi syndrome (frequency one in 10 000 to 25 000) and Angelman syndrome (exact frequency unknown) both result from the ‘microdeletion’ of the band 15q11–13 although they have different phenotypes depending upon the parental origin of the deletion. In Prader-Willi syndrome the deletion occurs invariably on the chromosome 15 inherited from the father whereas in Angelman syndrome the deletion occurs almost exclusively on the chromosome 15 from the mother. Hence inheritance of paternal and maternal copies of this region of chromosome 15 is important for normal development, a phenomenon known as genetic imprinting. People with Prader-Willi syndrome are mentally retarded, have small external genitalia and characteristic facial features. Babies with Prader-Willi syndrome have a poor sucking reflex hence feeding is difficult and results in weakness and stunted growth. Strangely, children with Prader-Willi syndrome become compulsive eaters at five to six years of age and suffer obesity and its related health problems . If untreated, afflicted individuals may feed themselves to death.
Angelman syndrome is characterized by developmental delay, absence of speech, jerky movements, paroxysms of inappropriate laughter and characteristic facial features which differ from those of Prader-Willi syndrome.
Many other portions of the genome are also subject to genetic imprinting although the precise mechanisms controlling whether the paternal or maternal copies of a gene are expressed are not fully understood.
DUPLICATIONS
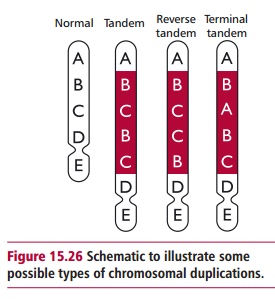
Chromosomal mutations that result in the doubling of a part of a chromosome are called duplications. The size of the duplicated segment varies enormously and duplicated segments may occur in a tandem configuration, that is, adjacent to each other or in different locations in the genome (Figure 15.26). Duplications can result in gene redundancy and may produce phenotypic variation, since there are now two copies of the gene and one copy may mutate independently of the other. This is thought to be a significant source of genetic variability during evolution. For example, gene duplications have been essential in the evolution of multigene families. These are groups of several genes whose products are similar in structure or functions. The genes for globins are a particularly well studied multigene family. Hemoglobins are tetrameric proteins consisting of two pairs of differing polypeptides each with an attached heme group able to bind and release a dioxygen molecule. Human individuals produce different hemoglobins at different stages during their lives as described. Each of the globin polypeptides has slightly different primary structures, but consist of the α types that are found as a cluster on chromosome 16 and the β types clustered on chromosome 11. It is thought that each group of genes evolved from one original ancestral gene that underwent duplications and subsequent sequence divergence.
INVERSIONS
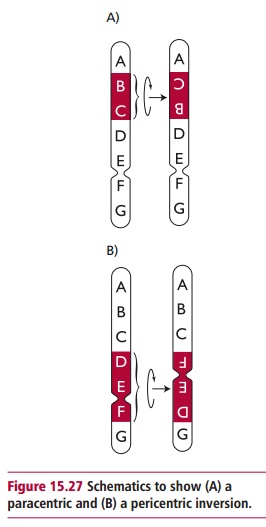
Figure 15.27 illustrates how inversions might arise. A chromosomal loopforms before fractures occur at two places on the chromosome. The insertion of the inverted segment at the newly created sticky ends and their subsequent joining within the chromosome completes the inversion. There are two types of inversions: paracentric inversions do not include the centromeres whereas pericentric inversions do. Genetic material is not lost during inversions although there can be clinical problems when fractures occur within genes or within regions that control gene expression. The meiotic consequences of a chromosomal inversion depend on the type of inversion encountered and the resulting gametes may be nonviable leading to reduced fertility.
TRANSLOCATIONS
Numerous translocations occur in the human population. The simplest kinds are intrachromosomal translocations that move part of a chromosome to a different position within the same chromosome. Interchromosomal translocations transfer part of a chromosome to a nonhomologous chromosome (Figure 15.28). Reciprocal translocation involves an interchromosomal translocation between two nonhomologous chromosomes. The least complex way for this event to occur is for two nonhomologous chromosome arms to come close to each other so that an exchange is facilitated.
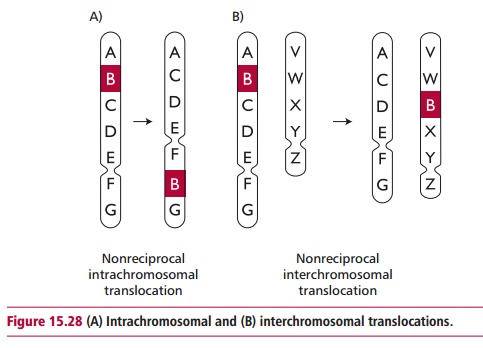
Homologues that are heterozygous for a reciprocal translocation undergo unorthodox synapsis during meiosis and pairing results in mitotic figures with a cross-like configuration. These chromosomes produce genetically unbalanced gametes and often result in reduced fertility. As few as 50% of the progeny of parents that are heterozygous for a reciprocal translocation survive, a condition known as semisterility. In humans, translocation can result in variations from the normal diploid number of chromosomes leading to a variety of birth defects. Translocations may transfer a gene to a region of a chromosome that is more transcriptionally active. This can lead to the development of some forms of cancer .
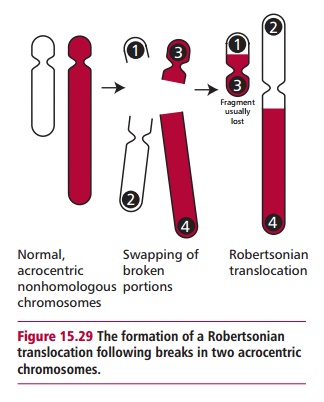
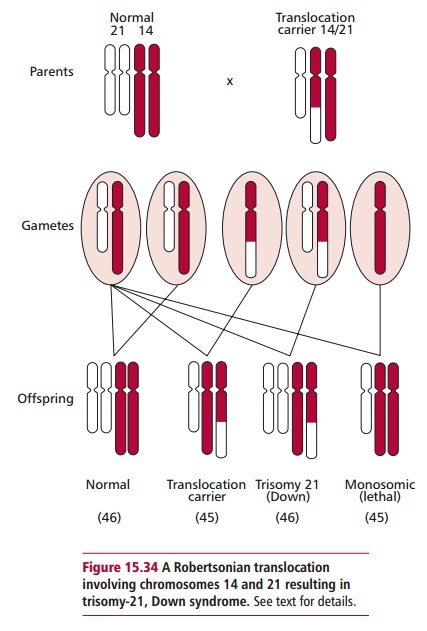
A common type of translocation involves breaks at the extreme ends of the short arms of two nonhomologous acrocentric chromosomes (Figures 15.29 and 15.34). The small fragments produced are lost but the larger ones fuse together at their centromeric regions. This type of translocation produces a new, large submetacentric or metacentric chromosome and is often called a Robertsonian translocation.
Related Topics