Chapter: Biology of Disease: Genetic Diseases
Chromosomes and the Human Karyotype
CHROMOSOMES AND THE HUMAN KARYOTYPE
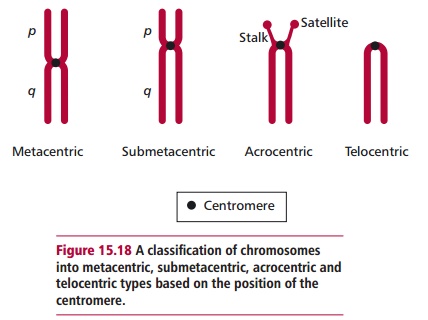
Interphase chromosomes are present as extended structures and cannot be seen with the light microscope. At the onset of cell division, both mitotic and meiotic, the chromosomes condense to form compact structures, referred to as mitotic figures. All chromosomes have a narrowed region called the centromere that divides the chromosome into two portions and allows them to be classified as metacentric, submetacentric, acrocentric or telocentric as shown in Figure 15.18. When the chromosome is divided into two unequal lengths, the shorter is called the p arm and the longer the q arm. When stained with Giemsa stain (Figure 15.19), most arms are divided into two or more regions by prominent bands and each region is further subdivided into subbands that can be numbered unambiguously. For example, band Xp21.2 is to be found on the p arm of the X chromosome in region 2, band 1, subband 2.

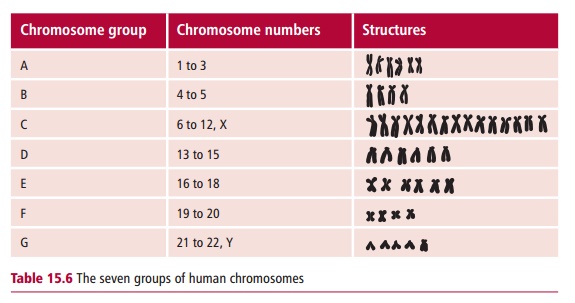
A karyotype is the characteristic number, size and shape of chromosomes of a species. A karyogram is a photographic representation of these chromosomes stained and arranged in order, as for example in Figures 15.2 and 15.20. An idiogram is a diagrammatic representation or interpretive drawing of thechromosomes based on the physical features seen in the karyogram. The karyotype of normal humans is 46. Human autosomal chromosomes are divided into seven groups (Table 15.6) on the basis of their sizes and the positions of their centromeres.

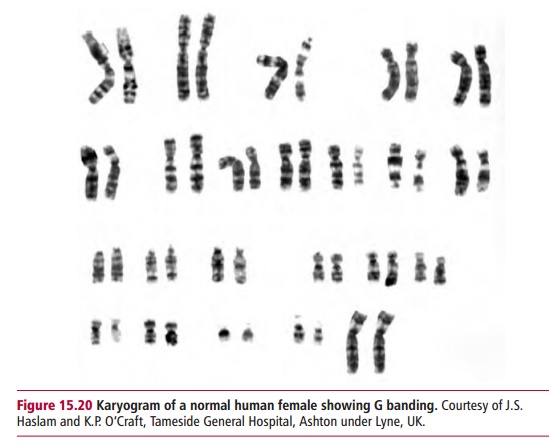
Cytogenetics is the microscopic study of chromosomes. Small lymphocytesisolated from a blood sample or cells obtained by amniocentesis or chorionic villus sampling are stimulated to divide by treatment with the plant lectin, phytohemagglutinin (PHA) and mitosis is then arrested at metaphase by an inhibitor such as colchicine. Metaphase chromosomes are visible by microscopy when stained in one of several different ways to allow their accurate identification when examined by microscopy. For routine karyotyping, Giemsa (G) staining is usually the preferred procedure since this produces a pattern of alternating dark and light bands characteristic for each pair of chromosomes. These patterns reflect differences in the detailed
structure of each chromosome. Karyotyping can be performed on white cells from whole blood as described above or from amniotic fluid, which contains cells from the developing fetus. When a patient’s chromosomes are examined using a microscope, it is possible to identify aberrations in chromosome number and structure.
The phenotype of a patient with a chromosomal disorder depends on the nature of their chromosomal defect. The first human chromosomal disorder was discovered in 1959 when three copies of chromosome 21 were found to be associated with Down syndrome. The development of chromosomal banding in 1970 has markedly increased the ability to resolve small chromosomal aberrations.
FRAGILE SITES
When human cells are grown in culture, some of the chromosomes in cells derived from certain individuals fail to stain in particular regions, giving the appearance of a gap. These sites are known as fragile sites, since they are susceptible to breakage when the cells are cultured in the absence of certain chemicals such as folic acid, which is normally present in the culture medium. More than 80 such sites have been identified since they were first discovered in 1965. The cause of the fragility at these sites is not known with certainty but they may represent regions where the DNA has been incompletely replicated.
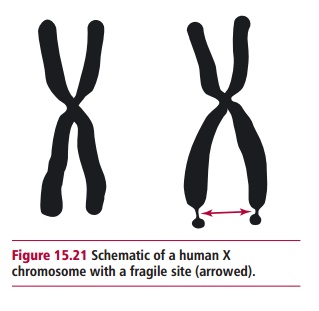
Almost all studies on fragile sites have been carried out in vitro on cells halted in mitosis. Initially they were not considered to be clinically relevant and, indeed, most fragile sites do not appear to be associated with any clinical syndrome. However, a strong association has been shown to exist between a form of mental retardation called fragile X or Martin-Bell syndrome and a fragile site on the X chromosome at position Xq27.3 (Figure 15.21) associated with the FMR1 gene. It is a dominant trait but fortunately fails to be fully expressed (incomplete penetrance) in many individuals. However, it is the commonest cause of mental retardation and has been estimated to affect one in about 4100 males in the UK. Most suffer mental retardation to the point that they are unable to live an independent life, and have a distinct physical appearance, including long, narrow faces with protruding chins, enlarged ears, and increased testicular size particularly after puberty. The syndrome also affects about one in 8000 females, who tend to suffer milder forms of retardation. Most humans carry a stable version of FMR1 which has about 30 CGG repeats. Individuals who have genes with about 45 to about 55 CGG repeats are in a gray zone; they do not have fragile X syndrome and, while they are likely to pass on a stable gene to their children, they have an increased chance of having children with a larger number of CGG repeats. People with about 55 to about 200 are said to have a premutation since although they generally have few or lack symptoms of fragile X syndrome, they can have children with more than 200 CGG repeats. This is the full mutation that initiates inappropriate methylation of cytosine bases and unfortunately can lead to the full blown syndrome.
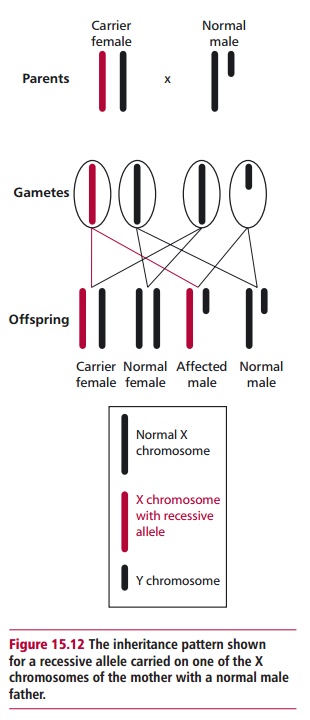
The FMR1 gene is carried on the X chromosome and shows Mendelian patterns of inheritance for X-linked disorders (Figure 15.12), although if it reaches the premutation stage it has a high probability of mutating (by repeat amplification) from one generation to the next. However, amplification of the CGG repeats can only occur in females, not males. Thus although there may be no family history of fragile X syndrome, it can suddenly appear in a number of offspring. The patterns of inheritance of fragile X syndrome are also somewhat complicated compared with a disorder like phenylketonuria, described. Given that FMR1 is on the X chromosome, a father cannot pass on any form of it to male offspring. However, daughters generally receive the paternal type. For example, almost all males with the stable version generally have daughters with the stable version. However, males with premutations, who are generally phenotypically normal and called normal transmitting males, have premutation type daughters. This inheritance can have severe consequences for any male grandchild as explained below. Most full mutation males do not have children. Those few who do would give the full mutated version to their daughters but surprisingly the daughters only express the premutation. Hence the father is passing on a reduced number of CGG repeats presumably because there are protected cells in the testes that never expand to the full mutation or a reduction in the repeat number occurs in some male reproductive cells. This means that all females who do have the full mutation must have received it from their mothers since they cannot receive it from their fathers.
Females have two X chromosomes and every child, male or female, has an equal, random chance of receiving one or the other of them. A female who has a copy of the premutation from her normal transmitting father can pass it on to her children. Most daughters who receive the premutation will show an increase in repeat number compared with their mother and, while most will show only the premutation, others will express the full mutation. Sons who inherit an X chromosome from a mother carrying a premutation are the principal group affected by fragile X syndrome since they are much more likely than females to have amplification to the full mutation. The probability of the full mutation is dependent upon the mother; those at the lower end of the premutation range, about 56–70 repeats, are less likely to have a son with the full mutation than those at the higher end with more than 100 repeats.
All males with the full mutation will experience significant symptoms. Some females with the full mutation will have symptoms of fragile X but, in general, the severity is less. Finally, there are individuals who cannot be assigned to these categories but have cells that vary regarding repeat size or the extent of methylation. The severity of their symptoms depends on the proportion of cells affected and the tissues involved.
The phenomenon of trinucleotide repeats is seen in several other human disorders. For example, a fragile site on chromosome 3 containing the gene FHIT (fragile histidine triad) is often altered in cells from tumors of patientswith lung cancer . Huntington disease, myotonic dystrophy and spinobulbar muscular atrophy or Kennedy disease are also associated with trinucleotide amplifications, although they differ from fragile X syndrome in that the amplification can occur in both sexes at each generation and is not associated with chromosome fragility. However, they are similar in that a threshold number of triplet repeats must be exceeded before symptoms of the disease appear.
Related Topics