Chapter: Medicine and surgery: Haematology and clinical Immunology
Acute lymphoblastic leukaemia (ALL) - Acute leukaemia
Acute leukaemia
Acute lymphoblastic leukaemia (ALL)
Definition
Clonal proliferation of lymphoid progenitor cells.
Incidence
85% of all childhood leukaemias.
Age
Predominantly seen in childhood with a peak incidence at 5 years. In adults incidence increases with advancing age.
Sex
M = F
Aetiology
ALL is more common in Down’s syndrome and in syndromes involving chromosomal instability like Fanconi anaemia. Risk factors include exposure to excessive radiation and benzene.
Pathophysiology
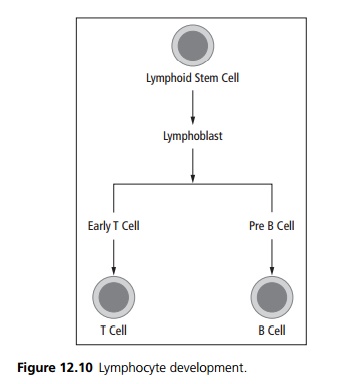
In acute leukaemias there is replacement of the normal bone marrow progenitor cells by blast cells, resulting in marrow failure. Acute lymphoblastic leukaemia arises from the lymphoid side of the haemopoetic system (see Fig. 12.10). It can be subdivided into:
Common ALL arising from the common progenitors. Pre B/B cell ALL.
Pre T/T cell ALL (50% present with a thymic mass). The blast cells of ALL tend to form reservoirs within the testes and CNS.
Clinical features
Often there is an insidious onset of anorexia, malaise and lethargy due to anaemia. There is often a history of recurrent infections and/or easy bruising and mucosal bleeding. Other presentations include lymph node en-largement, bone and joint pain and symptoms of raised intra cranial pressure. On examination there may be pallor, bruising, hepatosplenomegaly, lymphadenopathy,
papilloedema, cranial nerve palsies, testicular enlargement and occasionally superior vena caval obstruction.
Microscopy
The normal marrow is replaced by abnormal monotonous leukaemic cells of the lymphoid cell line. The leukaemia is typed by cytochemical staining and monoclonal antibodies to look for cell surface markers. The FAB (French/American/British) classification is based on morphology (FAB L1- homogenous population of small cells, FAB L2 heterogeneous population of cells, FAB L3 rare, cells similar to those in Burkitt’s lymphoma).
Investigations
Lymphoblasts are seen in a peripheral blood film. Full blood count shows a low haemoglobin, variable white count, low platelet count. Bone marrow aspiration shows increased cellularity with a high percentage of blast cells. Bone marrow cytogenetics and immunophenotyping is used to type the ALL and look for prognostic indicators.
Management
The current treatment protocol is UKALL-12. Which consists of:
· Induction: Phase 1 involves intravenous chemotherapy (daunorubicin and vincristine) oral prednisolone and intramuscular L-asparginase. Phase 2 involves intravenous chemotherapy (cyclophosphamide and cytosine) with oral 6-mercaptopurine.
· Intensification: This involves intravenous metho-trexate and folinic acid, with intramuscular L-asparginase.
· Consolidation: This involves several cycles of chemotherapy at lower doses.
· Maintenance: Therapy consists of 18 months of oral cytotoxic therapy with intravenous vincristine given every 3 months.
CNS treatment involves a combination of intrathecal methotrexate and cytosine and cranial irradiation.
Supportive treatment: Cytotoxic therapy and the leukaemia itself depresses normal bone marrow func-tion and causes a pancytopenia with resulting infection, anaemia and bleeding. Supportive therapy includes red cell concentrates, platelet transfusions, broad-spectrum antibiotics and prophylaxis for pneumocystic pneumonia.
The role of early bone marrow and haemopoetic stem cell transplantation is currently under investigation.
Prognosis
Prognosis is related to age, subtype and inversely proportional to the peripheral blast count. Over 90% of children respond to treatment, the rarer cases occurring in adults carry a worse prognosis.
Related Topics