Chapter: Medicine and surgery: Haematology and clinical Immunology
Sickle cell anaemia
Sickle cell anaemia
Definition
Autosomal recessive condition in which there is abnormal structure of the globin chain.
Incidence
Most common haemoglobinopathy.
Age
As HbF synthesis is normal, it presents at 6 months.
Sex
M = F
Geography
Occurs most frequently in Africa, Middle East, India and the Mediterranean.
Aetiology
A point mutation on chromosome 11 results in a substitution valine for glutamine at the sixth codon on the β globin chain to form haemoglobin (Hb)S. Infection, dehydration, hypoxia and cold may precipitate a sickle crisis. Classical sickle cell anaemia is a result of the presence of two mutations, i.e. HbSS, other combinations that produce sickle cell anaemia include Sβ thal, SD, SE and SC variants.
Pathophysiology
HbS molecules, when deoxygenated tend to aggregate into rigid polymers. The red blood cells become inflexible and sickle shaped and become trapped in the microcirculation, especially within bones, resulting in microvessel occlusion.
Clinical features
Sickle cell trait (the carrier state) is asymptomatic, but offers protection against falciparum malaria. Sickle cell anaemia is a clinical spectrum ranging from asymptomatic to severe haemolytic anaemia and recurrent sickle crises. Painful vascular occlusive crises typically produce symptoms of bone pain and pleuritic chest pain with a lowgrade fever. Severe occlusive crises may result in seizures and hemiparesis due to cerebral vessel occlusion, haematuria due to occlusion of the renal microcirculation, priapism, splenic infarcts and liver damage. Other patterns of crisis:
· Acute sequestration (pooling of blood in liver and spleen) requires transfusion for apparent hypovolaemia.
· Pulmonary infarction may occur in association with pneumonitis termed acute chest syndrome. Complications
Patients have a susceptibility to infections including streptococcal infections and osteomyelitis often due to salmonella. Avascular necrosis particularly of the femoral head may occur. Patients may develop renal failure due to recurrent renal damage and papillary necrosis. Retinal detachment and proliferative retinopathy may result in blindness. See also complications of haemolytic anaemia .
Investigations
· Full blood count shows anaemia. Blood film shows a high reticulocyte count and sickle shaped red blood cells.
· Sickle screening tests use a reducing solution, which causes HbS to precipitate. They are used as a first line test and for screening preoperatively.
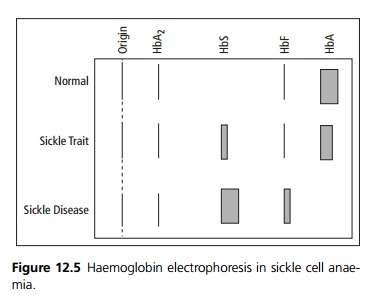
· Haemoglobin electrophoresis is used to diagnose both sickle cell trait and sickle cell disease (see Fig. 12.5).
· X-ray of the tubular bones may show destruction and medullary sclerosis together with periosteal bone formation.
Management
Treatment is largely symptomatic with prophylactic antibiotics, folic acid and pneumococcal vaccination. Management of a painful crisis includes oxygenation, adequate hydration and analgesia. Any associated bacterial infection should be treated with antibiotics. Acute sequestration requires blood transfusion, as patients become shocked. Severe crises such as priapism, acute chest syndrome or cerebral infarction require exchange blood transfusions to remove sickle cells. Transfusions may also be indicated in patients with regular severe crises and preoperatively.
Prognosis
There is marked variation in the severity of the condition, some patients have a relatively normal life span with few complications. In the most severe cases patients die from severe infarctions or sequestration.
Related Topics