Chapter: Medicine and surgery: Haematology and clinical Immunology
β-Thalassaemia
β-Thalassaemia
Definition
Inherited haemoglobinopathy with defective synthesis of the β-globulin chain.
Incidence
In the Mediterranean there is a carrier rate of 10–15%.
Age
Congenital
Sex
No sexual preponderance.
Geography
Most common in Asian and Mediterranean races.
Aetiology/pathophysiology
β-Thalassaemia results from point mutation in the β globin gene; over 100 defects have been characterised. These mutations may result in no β chain production (β0) or very reduced production (β+ ).
· Patients homozygous for β0 or β+ have very little or no production of β globin and have the clinical picture of β-thalassaemia major.
· Patients heterozygous for β0 or β+ have reduced β chain production and have a milder clinical picture termed β-thalassaemia minor.
Excess α chains precipitate in the red blood cells or combine with δ resulting in increased HbA2, and γ resulting in increased levels of fetal haemoglobin (HbF).
· If there are defects in both β and δ genes, patients have thalassaemia intermedia (homozygous) or thalassaemia minor (heterozygous).
· If there are defects in β, γ and δ, this results neonatal haemolysis and thalassaemia minor in heterozygous patients. Homozygous combined β, γ and δ are incompatible with life.
Clinical features
Thalassaemia minor/trait is asymptomatic with a mild hypochromic microcytic anaemia.
Thalassaemia intermedia causes symptomatic moderate anaemia with splenomegaly. Thalassaemia major presents in infancy with failure to thrive and recurrent infections. At 6 months the production of fetal haemoglobin ceases and the patient becomes symptomatic with a severe anaemia. Extramedullary haemopoesis causes hepatosplenomegaly, maxillary overgrowth and trabeculation on bone X-rays.
Investigations
Full blood count shows anaemia with a microcytic hypochromic appearance. The reticulocyte count is raised and there are nucleated red cells.
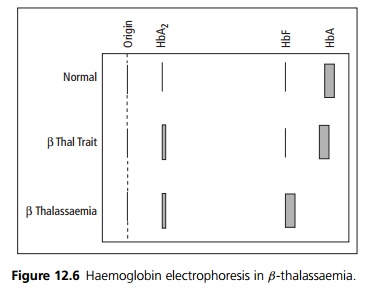
Haemoglobin electrophoresis with quantification of HbA2 is diagnostic (see Fig. 12.6).
Management
Thalassaemia minor does not require treatment; however, iron supplements should be avoided unless co-existent iron deficiency has been demonstrated. The partners of women with thalassaemia minor should be screened to allow appropriate genetic counselling.
Thalassaemia intermedia may require treatment for worsening anaemia or complications of haemolysis or extramedullary haemopoeisis.
Thalassaemia major and symptomatic thalassaemia intermedia are treated by regular blood transfusions to maintain a haemoglobin above 10 g/dL. This aims to suppress ineffective erythropoesis and prevent bony deformity, while allowing normal growth and development. Iron overload is prevented by the use of the chelating agent desferrioxamine, which is administered intravenously or by subcutaneous infusion. Splenectomy should be considered in patients over 6 years with high transfusion requirements. Bone marrow transplantation has been used successfully in young patients with severe β-thalassaemia major. Other treatments under investigation include gene therapy and drugs to maintain the production of fetal haemoglobin.
Related Topics