Chapter: Basic & Clinical Pharmacology : Antiviral Agents
Nucleoside & Nucleotide Reverse Transcriptase Inhibitors
NUCLEOSIDE & NUCLEOTIDE
REVERSE TRANSCRIPTASE INHIBITORS
The NRTIs act by competitive inhibition of HIV-1 reverse tran-scriptase; incorporation into the growing viral DNA chain causes premature chain termination due to inhibition of binding with the incoming nucleotide (Figure 49–4). Each agent requires intra-cytoplasmic activation via phosphorylation by cellular enzymes to the triphosphate form.
Typical
resistance mutations include M184V, L74V, D67N, and M41L. Lamivudine or
emtricitabine therapy tends to select rapidly for the M184V mutation in
regimens that are not fully suppressive. While the M184V mutation confers
reduced suscep-tibility to abacavir, didanosine, and zalcitabine, its presence
may restore phenotypic susceptibility to zidovudine. The K65R muta-tion is
associated with reduced susceptibility to tenofovir, abacavir, lamivudine, and
emtricitabine.
All NRTIs may be
associated with mitochondrial toxicity, prob-ably owing to inhibition of
mitochondrial DNA polymerase gamma. Less commonly, lactic acidosis with hepatic
steatosis may occur, which can be fatal. NRTI treatment should be suspended in
the setting of rapidly rising aminotransferase levels, progressive
hepatomegaly, or metabolic acidosis of unknown cause. The thymi-dine analogs
zidovudine and stavudine may be particularly associ-ated with dyslipidemia and
insulin resistance. Also, some evidence suggests an increased risk of
myocardial infarction in patients receiving abacavir or didanosine; this bears
further investigation.
ABACAVIR
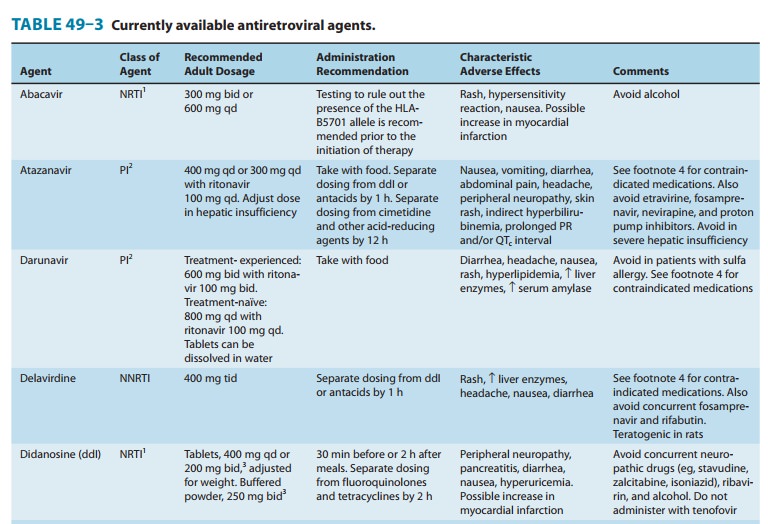
Abacavir is a guanosine analog (Figure 49–2) that is well absorbed following oral administration (83%) and is unaffected by food. The serum half-life is 1.5 hours. The drug undergoes hepatic glucuronidation and carboxylation. Cerebrospinal fluid levels are approximately one third those of plasma.Abacavir is often co-administered with lamivudine, and a once-daily, fixed-dose combination formulation is available. Abacavir is also available in a fixed-dose combination with lamivudine and zidovudine.High-level resistance to abacavir appears to require at least two or three concomitant mutations and thus tends to develop slowly.Hypersensitivity reactions, occasionally fatal, have been reported in up to 8% of patients receiving abacavir and may be more severe in association with once-daily dosing. Symptoms, which generally occur within the first 6 weeks of therapy, include fever, fatigue, nausea, vomiting, diarrhea, and abdominal pain.
Respiratory symptoms
such as dyspnea, pharyngitis, and cough may also be present, and skin rash
occurs in about 50% of patients. The laboratory abnormalities of a mildly
elevated serum aminotransferase or creatine kinase level may be present but are
nonspecific. Although the syndrome tends to resolve quickly with
discontinuation of medication, rechallenge with abacavir results in return of
symptoms within hours and may be fatal. Testing for the HLA-B5701 allele before
initiation of abacavir therapy is recom-mended to identify patients with an
increased risk for an abacavir-associated hypersensitivity reaction. Although
the positive predictive value of this test is only about 50%, it has a negative
predictive value of approximately 100%.
Other potential
adverse events are rash, fever, nausea, vomiting, diarrhea, headache, dyspnea,
fatigue, and pancreatitis (rare). Abacavir should be used cautiously in
patients with existing cardiac risk factors due to a possible increased risk of
myocardial events. Since abacavir may lower methadone levels, patients
receiving these two agents concurrently should be monitored for signs of opioid
withdrawal and may require an increased dose of methadone.
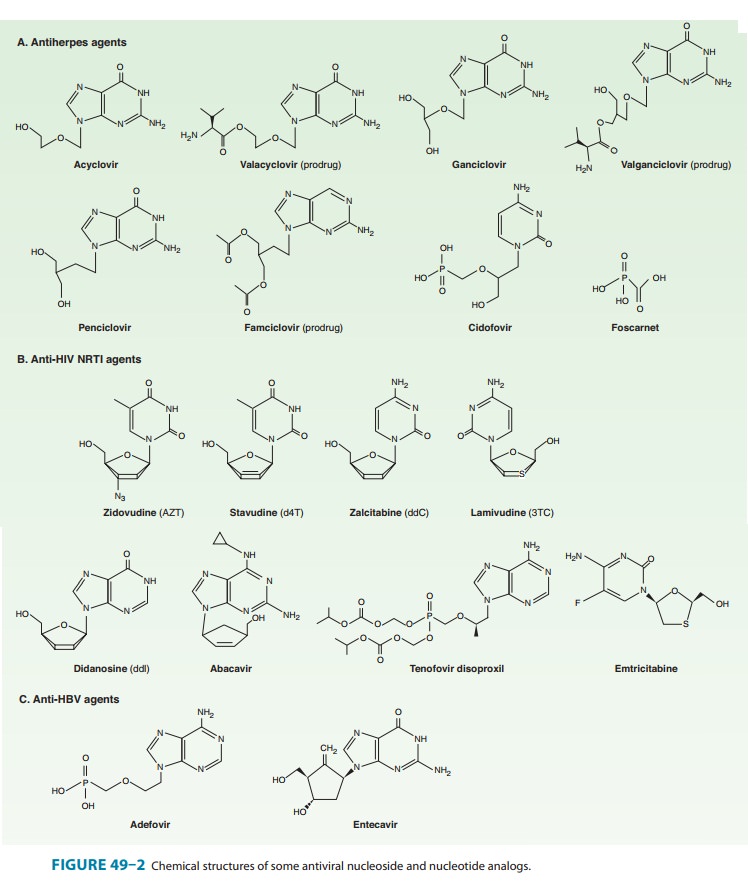
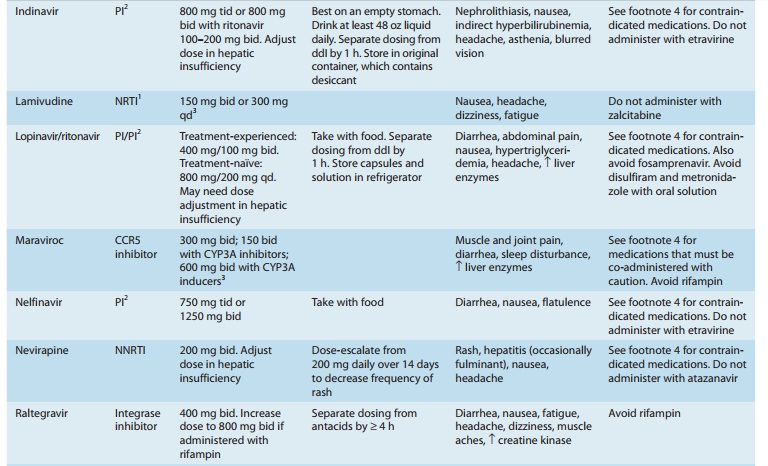
DIDANOSINE
Didanosine (ddI) is a
synthetic analog of deoxyadenosine (Figure 49–2). Oral bioavailability is
approximately 40%; dosing on an empty stomach is optimal, but buffered
formulations are neces-sary to prevent inactivation by gastric acid (Table
49–3). Cerebrospinal fluid concentrations of the drug are approximately 20% of
serum concentrations. Serum half-life is 1.5 hours, but the intracellular
half-life of the activated compound is as long as 20–24 hours. The drug is
eliminated by both cellular metabolism and renal excretionThe major clinical
toxicity associated with didanosine therapy is dose-dependent pancreatitis.
Other risk factors for pancreatitis
(eg,
alcohol abuse, hypertriglyceridemia) are relative contraindications, and
concurrent drugs with the potential to cause pancreatitis, includ-ing
zalcitabine, stavudine, ribavirin, and hydroxyurea, should be avoided (Table
49–3). The risk of peripheral distal sensory neuropa-thy, another potential
toxicity, may be increased with concurrent use of stavudine, isoniazid,
vincristine, or ribavirin. Other reported adverse effects include diarrhea
(particularly with the buffered formu-lation), hepatitis, esophageal
ulceration, cardiomyopathy, central nervous system toxicity (headache,
irritability, insomnia), and hyper-triglyceridemia. Previously asymptomatic
hyperuricemia may precipi-tate attacks of gout in susceptible individuals;
concurrent use of allopurinol may increase levels of didanosine. Reports of
retinal changes and optic neuritis in patients receiving didanosine,
particularly in adults receiving high doses and in children, mandate periodic
retinal examinations. Lipoatrophy appears to be more common in patients
receiving didanosine or other thymidine analogs. As with abacavir, didanosine
should be used cautiously in patients with cardiac risk fac-tors due to a
possibly increased risk of myocardial infarction.
The
buffer in didanosine tablets and powder interferes with absorption of
indinavir, delavirdine, atazanavir, dapsone, itracon-azole, and fluoroquinolone
agents; therefore, administration should be separated in time. Serum levels of
didanosine are increased when co-administered with tenofovir or ganciclovir,
and are decreased by atazanavir, delavirdine, ritonavir, tipranavir, and
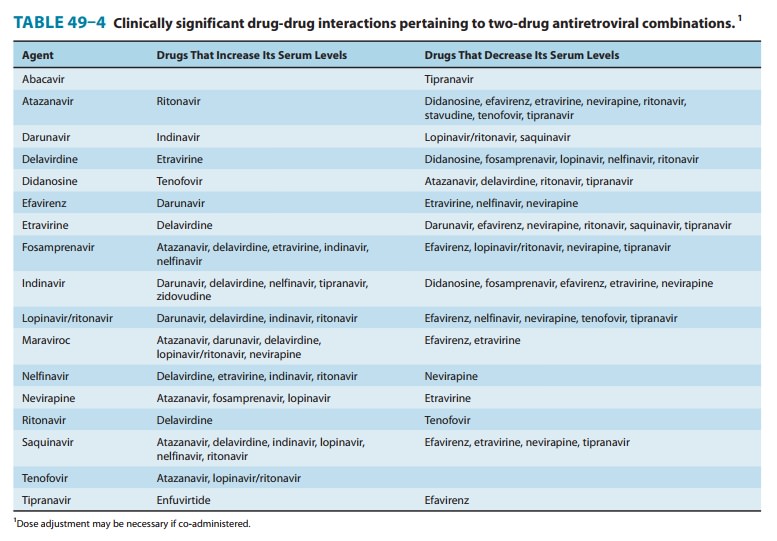
methadone (Table 49–4).
EMTRICITABINE
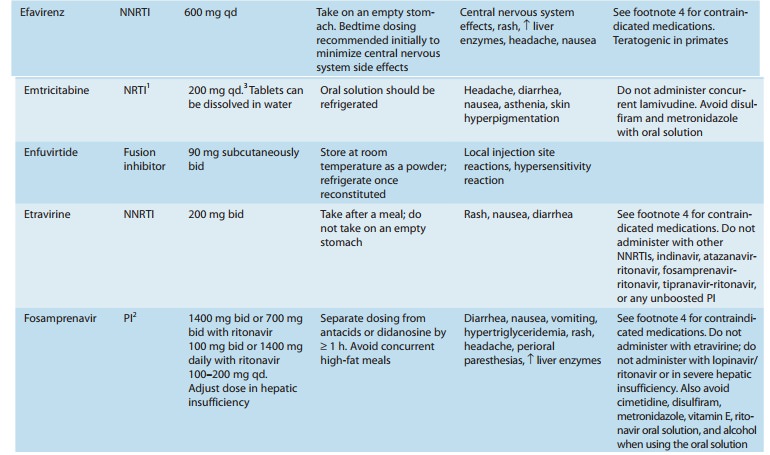
Emtricitabine (FTC) is a fluorinated analog of lamivudine with a long intracellular half-life (> 24 hours), allowing for once-daily dosing (Figure 49–2). Oral bioavailability of the capsules is 93% and is unaffected by food, but penetration into the cerebrospinal fluid is low. Elimination is by both glomerular filtration and active tubular secretion. The serum half-life is about 10 hours.
The
oral solution, which contains propylene glycol, is contraindi-cated in young
children, pregnant women, patients with renal or hepatic failure, and those
using metronidazole or disulfiram. Also, because of its activity against HBV,
patients co-infected with HIV and HBV should be closely monitored if treatment
with emtricitabine is interrupted or discontinued, owing to the likelihood of
hepatitis flare.
Emtricitabine
is often co-administered with tenofovir, and a once-daily, fixed-dose
combination formulation is available, both alone and in combination with
efavirenz. In a recent placebo-controlled study, use of emtricitabine and
tenofovir was effective as preexposure prophylaxis, reducing HIV acquisition in
men who have sex with men.
Like lamivudine, the M184V/I mutation is most frequently associated with emtricitabine use and may emerge rapidly in patients receiving regimens that are not fully suppressive. Because of their similar mechanisms of action and resistance profiles, the com-bination of lamivudine and emtricitabine is not recommended.
The most common
adverse effects observed in patients receiv-ing emtricitabine are headache,
diarrhea, nausea, and rash. In addition, hyperpigmentation of the palms or
soles may be observed (∼
3%), particularly in African-Americans (up to 13%). No drug-drug interactions
of note have been reported to date.
LAMIVUDINE
Lamivudine (3TC) is a
cytosine analog (Figure 49–2) with in vitro activity against HIV-1 that is
synergistic with a variety of antiret-roviral nucleoside analogs—including
zidovudine and stavudine— against both zidovudine-sensitive and
zidovudine-resistant HIV-1 strains. As with emtricitabine, lamivudine has
activity against HBV; therefore, discontinuation in patients that are
co-infected with HIV and HBV may be associated with a flare of hepatitis.
Oral
bioavailability exceeds 80% and is not food-dependent. In children, the average
cerebrospinal fluid:plasma ratio of lamivudine was 0.2. Serum half-life is 2.5
hours, whereas the intracellular half-life of the triphosphorylated compound is
11–14 hours. Most of the drug is eliminated unchanged in the urine.
Lamivudine
is often co-administered with abacavir, and a once-daily, fixed-dose
combination formulation is available. Lamivudine is also available in a
fixed-dose combination with zidovudine, either alone or in combination with
abacavir.
Lamivudine therapy
rapidly selects for the M184V mutation in regimens that are not fully
suppressive.Potential adverse effects are headache, dizziness,
insomnia, fatigue, dry mouth, and gastrointestinal discomfort, although these
are typically mild and infrequent. Lamivudine’s bioavail-ability increases when
it is co-administered with trimethoprim-sulfamethoxazole. Lamivudine and
zalcitabine may inhibit the intracellular phosphorylation of one another;
therefore, their concurrent use should be avoided if possible. Short-term
safety of lamivudine has been demonstrated for both mother and infant.
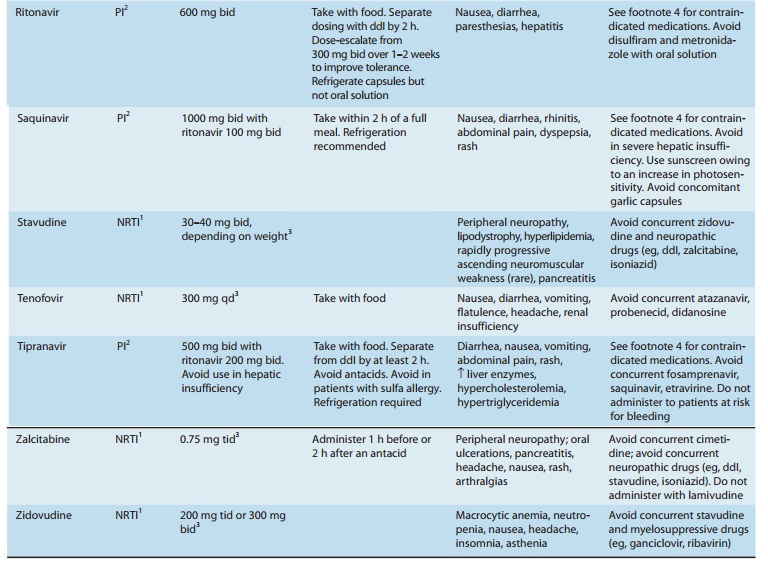
STAVUDINE
The
thymidine analog stavudine (d4T) (Figure 49–2) has high oral bioavailability
(86%) that is not food-dependent. The serum half-life is 1.1 hours, the
intracellular half-life is 3.0–3.5 hours, and mean cerebrospinal fluid
concentrations are 55% of those of plasma. Excretion is by active tubular
secretion and glomerular filtration.
The
major toxicity is a dose-related peripheral sensory neuropathy. The incidence
of neuropathy may be increased when stavudine is administered with other
neuropathy-inducing drugs such as didanosine, zalcitabine, vincristine,
isoniazid, or ribavirin, or in patients with advanced immunosuppression.
Symptoms typically resolve upon discontinuation of stavudine; in such cases, a
reduced dosage may be cautiously restarted. Other potential adverse effects are
pancreatitis, arthralgias, and elevation in serum aminotransferases. Lactic
acidosis with hepatic steatosis, as well as lipodystrophy, appear to occur more
frequently in patients receiv-ing stavudine than in those receiving other NRTI
agents. Moreover, because the co-administration of stavudine and didanos-ine
may increase the incidence of lactic acidosis and pancreatitis, concurrent use
should be avoided. This combination has been implicated in several deaths in
HIV-infected pregnant women. A rare adverse effect is a rapidly progressive
ascending neuromuscu-lar weakness. Since zidovudine may reduce the
phosphorylation of stavudine, these two drugs should not be used together.
There is no evidence of human teratogenicity in those taking stavudine.
TENOFOVIR
Tenofovir is an
acyclic nucleoside phosphonate (ie, nucleotide) analog of adenosine (Figure
49–2). Like the nucleoside analogs, tenofovir competitively inhibits HIV
reverse transcriptase and causes chain termination after incorporation into
DNA. However, only two rather than three intracellular phosphorylations are
required for active inhibition of DNA synthesis. Tenofovir is also approved for
the treatment of patients with HBV infection.
Tenofovir disoproxil
fumarate is a water-soluble prodrug of active tenofovir. The oral
bioavailability in fasted patients is approximately 25% and increases to 39%
after a high-fat meal. The prolonged serum (12–17 hours) and intracellular
half-lives allow once-daily dosing. Elimination occurs by both glomerular
filtration and active tubular secretion.
Tenofovir
is often co-administered with emtricitabine, and a once-daily, fixed-dose
combination formulation is available, either alone or in combination with
efavirenz. A recent placebo-controlled study found that use of emtricitabine
and tenofovir was effective as preexposure prophylaxis, reducing HIV
acquisi-tion in men who have sex with men. In another placebo-con-trolled
study, use of the experimental 1% tenofovir gel as a vaginal microbicide was
effective in decreasing the incidence of heterosexual HIV acquisition.
The
primary mutation associated with resistance to tenofovir is K65R. Gastrointestinal
complaints (eg, nausea, diarrhea, vomiting, flatulence) are the most common
adverse effects but rarely require discontinuation of therapy. Since tenofovir
is formulated with lactose, these may occur more frequently in patients with
lactose intolerance. Other potential adverse effects include headache and
asthenia. Tenofovir-associated proximal renal tubulopathy causes excessive
renal phosphate and calcium losses and 1-hydroxylation defects of vitamin D,
and preclinical studies in several animal spe-cies have demonstrated bone
toxicity (eg, osteomalacia). Monitoring of bone mineral density should be
considered with long-term use in those with risk factors for or with known
osteoporosis, as well as in children. Reduction of renal function over time, as
well as cases of acute renal failure and Fanconi’s syndrome, have been reported
in patients receiving tenofovir alone or in combination with emtricit-abine.
For this reason, tenofovir should be used with caution in patients at risk for
renal dysfunction. Tenofovir may compete with other drugs that are actively
secreted by the kidneys, such as cidofovir, acyclovir, and ganciclovir. Concurrent
use of atazanavir or lopinavir/ ritonavir may increase serum levels of
tenofovir (Table 49–4).
Tenofovir
is associated with decreased fetal growth and reduc-tion in fetal bone porosity
in monkeys. There is significant placental passage in humans.
ZALCITABINE
Zalcitabine (ddC) is a
cytosine analog with high oral bioavailabil-ity (87%) and a serum half-life of
1–2 hours (Figure 49–2). An intracellular half-life of 2.6 hours necessitates
thrice-daily dosing, which limits its usefulness. Plasma levels decrease by
25–39% when the drug is administered with food or antacids. The drug is
excreted renally. Cerebrospinal fluid concentrations are approxi-mately 20% of
those in the plasma.
Although a variety of
mutations associated with in vitro resis-tance to zalcitabine have been
described, phenotypic resistance appears to be rare.
Zalcitabine
therapy is associated with a dose-dependent periph-eral neuropathy that can be
treatment-limiting in 10–20% of patients but appears to be slowly reversible if
treatment is stopped promptly. The potential for causing peripheral neuropathy
consti-tutes a relative contraindication to use with other drugs that may cause
neuropathy, including stavudine, didanosine, isoniazid, vincristine, and
ribavirin. Decreased creatinine clearance or concur-rent use of potential
nephrotoxins (eg, amphotericin B, foscarnet, and aminoglycosides) may increase
the risk of zalcitabine neuropa-thy, as does more advanced immunosuppression.
The other major reported toxicity is oral and esophageal ulceration.
Pancreatitis occurs less frequently than with didanosine administration, but
co-administration of other drugs that cause pancreatitis may increase the
frequency of this adverse effect. Headache, nausea, rash, and arthralgias may
occur but tend to be mild or to resolve during therapy. Zalcitabine causes
thymic lymphoma in rodents, as well as hydrocephalus at high doses; clinical
relevance is unclear. The AUC of zalcitabine increases when co-administered
with probenecid or cimetidine, and bioavailability decreases with concurrent
antacids or metoclopramide. Lamivudine inhibits the phosphorylation of
zalcitabine in vitro, potentially interfering with its efficacy.
ZIDOVUDINE
Zidovudine (azidothymidine; AZT) is a
deoxythymidine analog (Figure 49–2) that is well absorbed (63%) and distributed
to most body tissues and fluids, including the cerebrospinal fluid, where drug
levels are 60–65% of those in serum. Although the serumhalf-life averages 1
hour, the intracellular half-life of the phospho-rylated compound is 3–4 hours,
allowing twice-daily dosing. Zidovudine is eliminated primarily by renal
excretion following glucuronidation in the liver.
Zidovudine is
available in a fixed-dose combination formulation with lamivudine, either alone
or in combination with abacavir.Zidovudine was the first antiretroviral agent
to be approved and has been well studied. The drug has been shown to decrease
the rate of clinical disease progression and prolong survival in HIV-infected
individuals. Efficacy has also been demonstrated in the treatment of
HIV-associated dementia and thrombocytope-nia. In pregnancy (Table 49–5), a
regimen of oral zidovudine beginning between 14 and 34 weeks of gestation,
intravenous zidovudine during labor, and zidovudine syrup to the neonate from
birth through 6 weeks of age has been shown to reduce the rate of vertical
(mother-to-newborn) transmission of HIV by up to 23%.

High-level
zidovudine resistance is generally seen in strains with three or more of the
five most common mutations: M41L, D67N, K70R, T215F, and K219Q. However, the
emergence of certain mutations that confer decreased susceptibility to one drug
(eg, L74V for didanosine and M184V for lamivudine) may enhance zidovudine
susceptibility in previously zidovudine-resistant strains. Withdrawal of
zidovudine may permit the rever-sion of zidovudine-resistant HIV-1 isolates to
the susceptible wild-type phenotype.The most common adverse effect of zidovudine is
myelosup-pression, resulting in macrocytic anemia (1–4%) or neutropenia (2–8%).
Gastrointestinal intolerance, headaches, and insomnia may occur but tend to
resolve during therapy. Lipoatrophy appears to be more common in patients
receiving zidovudine or other thymidine analogs. Less common toxicities include
throm-bocytopenia, hyperpigmentation of the nails, and myopathy. High doses can
cause anxiety, confusion, and tremulousness. Zidovudine causes vaginal
neoplasms in mice; however, no human cases of genital neoplasms have been
reported to date. Short-term safety has been demonstrated for both mother and
infant.
Increased serum levels
of zidovudine may occur with concomi-tant administration of probenecid,
phenytoin, methadone, flucon-azole, atovaquone, valproic acid, and lamivudine,
either through inhibition of first-pass metabolism or through decreased
clearance. Zidovudine may decrease phenytoin levels. Hematologic toxicity may
be increased during co-administration of other myelosuppres-sive drugs such as
ganciclovir, ribavirin, and cytotoxic agents. Combination regimens containing
zidovudine and stavudine should be avoided due to in vitro antagonism.
Related Topics