Chapter: Clinical Anesthesiology: Perioperative & Critical Care Medicine: Management of Patients with Fluid & Electrolyte Disturbances
Hypokalemia
HYPOKALEMIA
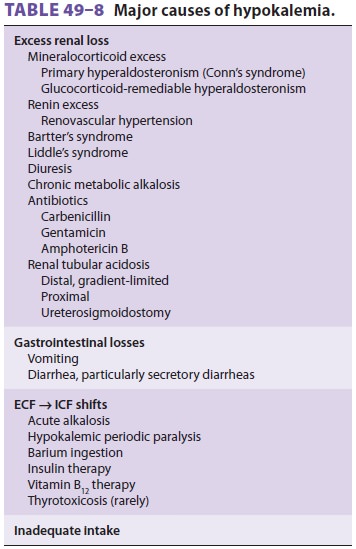
Hypokalemia, defined as plasma [K+] less than 3.5 mEq/L, can occur as a result of (1) an
intercom-partmental shift of K+ (see above), (2)
increased potassium loss, or (3) an inadequate potassium intake (Table 49–8). Plasma potassium
concentration typi-cally correlates poorly with the total potassium deficit. A
decrease in plasma [K+] from 4 mEq/L to 3
mEq/L usually represents a 100- to 200-mEq deficit, whereas plasma [K+] below 3 mEq/L can represent a deficit anywhere between 200 mEq and 400
mEq.
Hypokalemia due to the Intracellular Movement of Potassium
Hypokalemia due to the intracellular movement of potassium occurs with
alkalosis, insulin therapy, β2-adrenergic
agonists, and hypothermia and dur-ing attacks of hypokalemic periodic paralysis
(see above). Hypokalemia may also be seen following transfusion of previously
frozen red cells; these cells lose potassium in the preservation process and
take up potassium following reinfusion. Cel-lular K+ uptake by red blood cells (and
platelets) also accounts for the hypokalemia seen in patients recently treated
with folate or vitamin B12 for mega-loblastic anemia.
Hypokalemia due to Increased Potassium Losses
Excessive potassium losses are usually either
renal or gastrointestinal. Renal wasting of potassium is most commonly the
result of diuresis or enhanced mineralocorticoid activity. Other renal causes
include hypomagnesemia , renal tubular acidosis , ketoacidosis, salt-wasting
nephropathies, and some drug therapies (carbenicil-lin and amphotericin B).
Increased gastrointestinal loss of potassium is most commonly due to
nasogas-tric suctioning or to persistent vomiting or diarrhea. Other
gastrointestinal causes include losses from fis-tulae, laxative abuse, villous
adenomas, and pancre-atic tumors secreting vasoactive intestinal peptide.
Chronic
increased sweat formation occasionally causes hypokalemia, particularly when
potassium intake is limited. Dialysis with a low-potassium-containing dialysate
solution can also cause hypo-kalemia. Uremic patients may actually have a total
body potassium deficit (primarily intracellular) despite a normal or even high
plasma concentra-tion; the absence of hypokalemia in these instances is
probably due to an intercompartmental shift from the acidosis. Dialysis in
these patients unmasks the total body potassium deficit and often results in
hypokalemia.
Urinary [K+] less than 20 mEq/L is generally indicative of increased extrarenal
losses, whereas concentrations greater than 20 mEq/L suggest renal wasting of K+.
Hypokalemia due to Decreased Potassium Intake
Because of the kidney’s ability to decrease urinary potassium excretion to
as low as 5–20 mEq/L, marked reductions in potassium intake are required to
produce hypokalemia. Low potassium intakes, however, often accentuate the
effects of increased potassium losses.
Clinical Manifestations of Hypokalemia
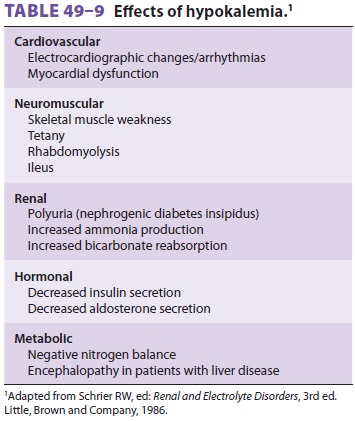
Hypokalemia can produce widespread organ dysfunc-tion (Table
49–9). Most patients are asymptomatic until plasma [K+] falls below 3 mEq/L.
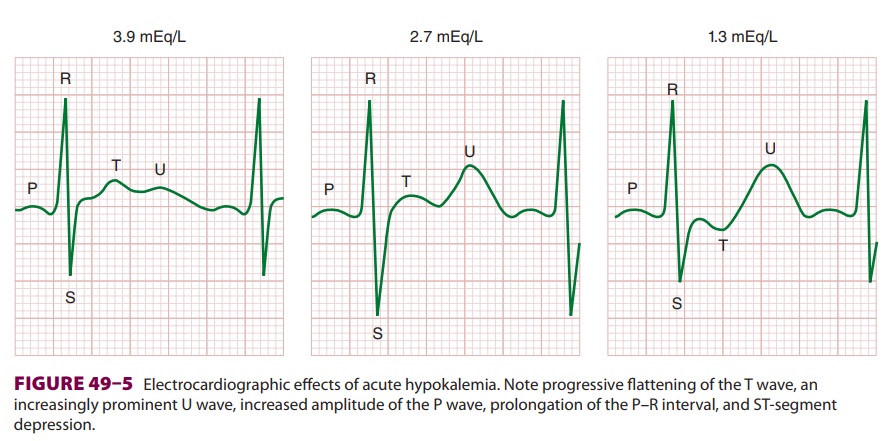
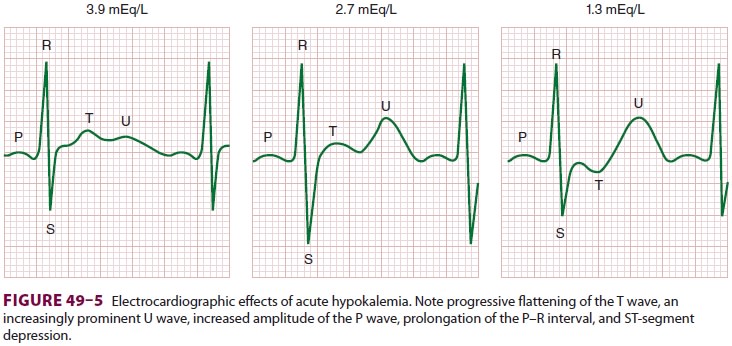
Cardiovascular effects are most prominent and include an abnormal ECG (Figure
49–5), arrhythmias, decreased cardiac contractility, and a labile arterial
blood pressure due to autonomic dysfunction. Chronic hypokalemia has also been reported
to cause myocardial fibrosis.
ECG manifestations are primarily due to
delayed ventricular repolarization and include T-wave flat-tening and inversion, an increasingly prominent U wave, ST-segment
depression, increased P-wave amplitude, and prolongation of the P–R interval.
Increased myocardial cell automaticity and delayed repolarization
promote both atrial and ventricular arrhythmias.
Neuromuscular effects of hypokalemia include skeletal muscle weakness,
flaccid paralysis, hypo-reflexia, muscle cramping, ileus, and, rarely,
rhab-domyolysis. Hypokalemia induced by diuretics is often associated with
metabolic alkalosis; as the kidneys absorb sodium to compensate for
intra-vascular volume depletion and in the presence of diuretic-induced
hypochloremia, bicarbonate is absorbed. The end result is hypokalemia and
hypo-chloremic metabolic alkalosis. Renal dysfunction is seen due to impaired
concentrating ability (resis-tance to ADH, resulting in polyuria) and increased
production of ammonia resulting in impairment of urinary acidification.
Increased ammonia pro-duction represents intracellular acidosis; hydrogen ions
move intracellularly to compensate for intra-cellular potassium losses. The
resulting metabolic alkalosis, together with increased ammonia pro-duction, can
precipitate encephalopathy in patients with advanced liver disease. Chronic
hypokalemia has been associated with renal fibrosis (tubulointer-stitial nephropathy).
Treatment of Hypokalemia
The treatment of hypokalemia depends on the presence and severity of any
associated organ dysfunction. Significant ECG changes such as ST-segment
changes or arrhythmias mandate con-tinuous ECG monitoring, particularly during
intravenous K+
replacement. Digoxin therapy—as well as the hypokalemia itself—sensitizes the
heart to changes in potassium ion concentration. Muscle strength should also be
periodically assessed in patients with weakness.
In most circumstances, the safest method by
which to correct a potassium deficit is oral replace-ment over several days
using a potassium chloride solution (60–80 mEq/d). Intravenous replacement of
potassium chloride is usuallybe reserved for patients with, or at risk for,
signifi-cant cardiac manifestations or severe muscle weakness. The goal of
intravenous therapy is to remove the patient from immediate danger, not to
correct the entire potassium deficit. Because of potassium’s irritative effect
on peripheral veins, peripheral intravenous replacement should not exceed 8
mEq/h. Dextrose-containing solutions should generally be avoided because the
resulting hyperglycemia and secondary insulin secretion may actually worsen the
low plasma [K+]. More rapid intravenous potassium replacement (10–20 mEq/h) requires
central venous adminis-tration and close monitoring of the ECG. Intrave-nous
replacement should generally not exceed 240 mEq/d. Potassium chloride is the
preferred potas-sium salt when a metabolic alkalosis is also present because it
also corrects the chloride deficit discussed above. Potassium bicarbonate or
equivalent (K acetate or K citrate) is preferable for patients with metabolic
acidosis. Potassium phosphate is a suit-able alternative with concomitant
hypophosphate-mia (diabetic ketoacidosis).
Anesthetic Considerations
Hypokalemia is a common preoperative finding. The decision to proceed
with elective surgery is often based on lower plasma [K+] limits somewhere between 3
and 3.5 mEq/L. The decision, how-ever, should also be based on the rate at
which the hypokalemia developed as well as the presence or absence of secondary
organ dysfunction. In general, chronic mild hypokalemia (3–3.5 mEq/L) without
ECG changes does not substantially increase anes-thetic risk. The latter may
not apply to patients receiving digoxin, who may be at increased risk of
developing digoxin toxicity from the hypokalemia; plasma [K +] values above 4 mEq/L are
desirable in such patients.
The intraoperative management of hypokale-mia requires vigilant ECG
monitoring. Intravenous potassium should be given if atrial or ventricular
arrhythmias develop. Glucose-free intravenous solutions should be used and
hyperventilation avoided to prevent further decreases in plasma [K +]. Increased sensitivity to
neuromuscular blockers (NMBs) may be seen; therefore dosages of NMBs should be
reduced 25–50%, and a nerve stimulator should be used to follow both the degree
of paralysis and the adequacy of reversal.
Related Topics