Chapter: Basic & Clinical Pharmacology : Antiviral Agents
Agents to Treat Herpes Simplex Virus HSV & Varicella Zoster Virus VZV Infections
AGENTS TO TREAT HERPES SIMPLEX VIRUS HSV & VARICELLA ZOSTER
VIRUS VZV INFECTIONS
Three oral nucleoside
analogs are licensed for the treatment of HSV and VZV infections: acyclovir,
valacyclovir, and famciclovir. They have similar mechanisms of action and
comparable indications for clinical use; all are well tolerated. Acyclovir has
been the most exten-sively studied; it was licensed first and is the only one
of the three that is available for intravenous use in the United States.
Comparative trials have demonstrated similar efficacies of these three agents
for the treatment of HSV but modest superiority of famciclovir and valacyclovir
for the treatment of herpes zoster infections
ACYCLOVIR
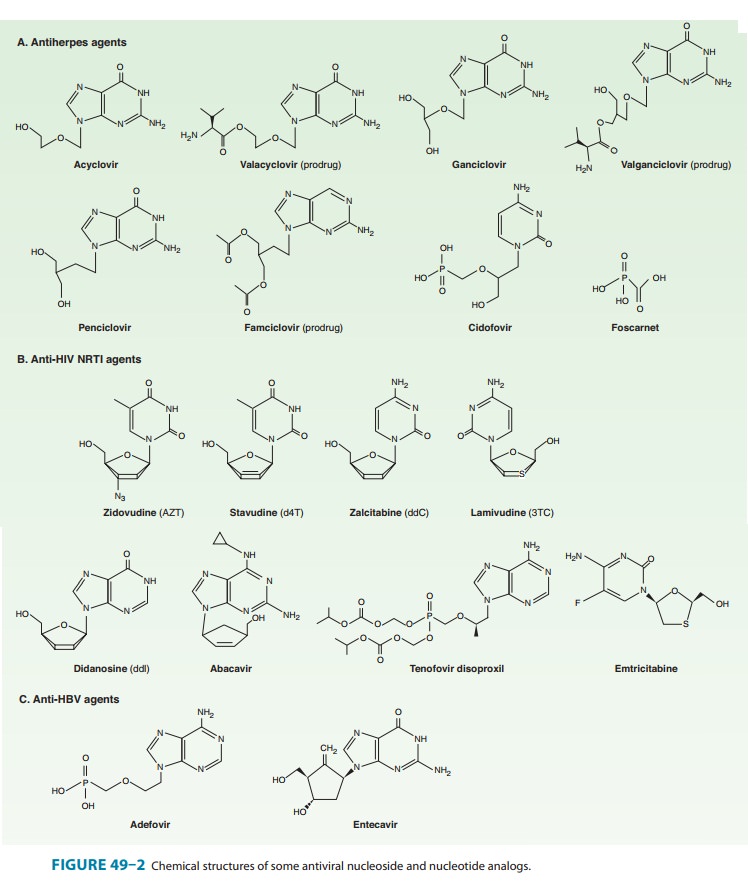
Acyclovir
(Figure 49–2) is an acyclic guanosine derivative with clinical activity against
HSV-1, HSV-2, and VZV, but it is approx-imately 10 times more potent against
HSV-1 and HSV-2 than against VZV. In vitro activity against Epstein-Barr virus
(EBV), cytomegalovirus (CMV), and human herpesvirus-6 (HHV-6) is present but
weaker.
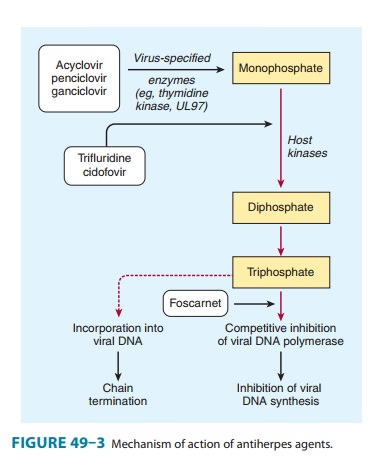
Acyclovir
requires three phosphorylation steps for activation. It is converted first to
the monophosphate derivative by the virus-specified thymidine kinase and then
to the di- and triphosphate compounds by host cell enzymes (Figure 49–3).
Because it requires the viral kinase for initial phosphorylation, acyclovir is
selectively activated—and the active metabolite accumulates— only in infected
cells. Acyclovir triphosphate inhibits viral DNA synthesis by two mechanisms:
competition with deoxyGTP for the viral DNA polymerase, resulting in binding to
the DNA tem-plate as an irreversible complex; and chain termination following
incorporation into the viral DNA.
The
bioavailability of oral acyclovir is low (15–20%) and is unaffected by food. An
intravenous formulation is available. Topical formulations produce high
concentrations in herpetic lesions, but systemic concentrations are
undetectable by this route.
Acyclovir is cleared primarily by glomerular filtration and tubular secretion. The half-life is 2.5–3 hours in patients with normal renal function and 20 hours in patients with anuria.
Acyclovir
diffuses readily into most tissues and body fluids. Cerebrospinal fluid concentrations are 20–50% of serum
values.
Oral
acyclovir has multiple uses. In first episodes of genital herpes, oral
acyclovir shortens the duration of symptoms by approximately 2 days, the time
to lesion healing by 4 days, and the duration of viral shedding by 7 days. In
recurrent genital herpes, the time course is shortened by 1–2 days. Treatment
of first-episode genital herpes does not alter the frequency or severity of
recurrent outbreaks. Long-term suppression with oral acyclovir in patients with
frequent recurrences of genital herpes decreases the
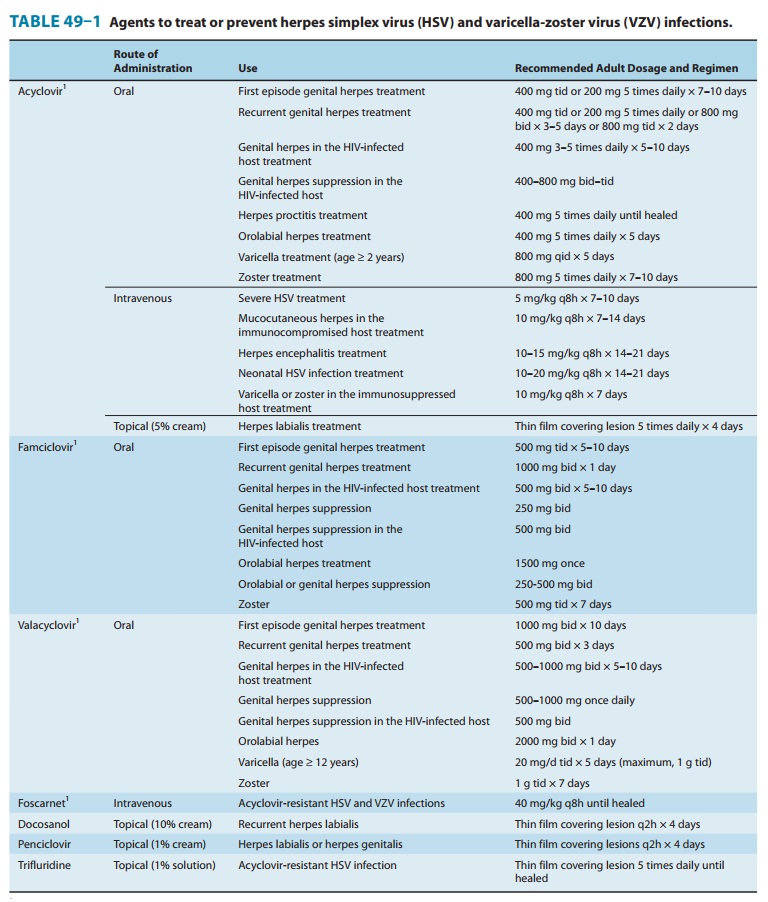
frequency
of symptomatic recurrences and of asymptomatic viral shedding, thus decreasing
the rate of sexual transmission. However, outbreaks may resume upon
discontinuation of suppressive acyclovir. Oral acyclovir is only modestly
beneficial in recurrent herpes labialis. In contrast, acyclovir therapy
significantly decreases the total number of lesions, duration of symptoms, and
viral shed-ding in patients with varicella (if begun within 24 hours after the
onset of rash) or cutaneous zoster (if begun within 72 hours). However, because
VZV is less susceptible to acyclovir than HSV, higher doses are required (Table
49–1). When given prophylacti-cally to patients undergoing organ
transplantation, oral or intrave-nous acyclovir prevents reactivation of HSV
infection. Evidence from recent clinical trials suggests that the use of daily
acyclovir (400 mg twice daily) may reduce the plasma viral load of HIV-1 and
the risk of HIV-associated disease progression in individuals dually infected
with HSV-2 and HIV-1.
Intravenous acyclovir
is the treatment of choice for herpes simplex encephalitis, neonatal HSV
infection, and serious HSV or VZV infections (Table 49–1). In immunocompromised
patients with VZV infection, intravenous acyclovir reduces the incidence of
cutaneous and visceral dissemination.
Topical acyclovir
cream is substantially less effective than oral therapy for primary HSV
infection. It is of no benefit in treating recurrent genital herpes.
Resistance
to acyclovir can develop in HSV or VZV through alteration in either the viral
thymidine kinase or the DNA polymerase, and clinically resistant infections
have been reported in immunocompromised hosts. Most clinical isolates are
resistant on the basis of deficient thymidine kinase activity and thus are
cross-resistant to valacyclovir, famciclovir, and ganciclovir. Agents such as
foscarnet, cidofovir, and trifluridine do not require activation by viral
thymidine kinase and thus have preserved activity against the most prevalent
acyclovir-resistant strains (Figure 49–3).
Acyclovir is generally well tolerated. Nausea, diarrhea, and headache have occasionally been reported. Intravenous infusion may be associated with reversible renal toxicity (ie, crystalline nephropathy or interstitial nephritis) or neurologic effects (eg, tremors, delirium, seizures). However, these are uncommon with adequate hydration and avoidance of rapid infusion rates. High doses of acyclovir cause chromosomal damage and testicular atrophy in rats, but there has been no evidence of teratogenicity, reduction in sperm production, or cytogenetic alterations in peripheral blood lymphocytes in patients receiving daily suppres-sion of genital herpes for more than 10 years. A recent study found no evidence of increased birth defects in 1150 infants who were exposed to acyclovir during the first trimester.
Concurrent use of
nephrotoxic agents may enhance the poten-tial for nephrotoxicity. Probenecid
and cimetidine decrease acyclo-vir clearance and increase exposure. Somnolence
and lethargy may occur in patients receiving concomitant zidovudine and
acyclovir.
VALACYCLOVIR
Valacyclovir
is the L-valyl ester of acyclovir (Figure 49–2). It is rapidly
converted to acyclovir after oral administration via first-pass enzymatic
hydrolysis in the liver and intestine, resulting in serum levels that are three
to five times greater than those achieved with oral acyclovir and approximate
those achieved with intrave-nous acyclovir. Oral bioavailability is 54–70%, and
cerebrospinal fluid levels are about 50% of those in serum. Elimination
half-life is 2.5–3.3 hours.
Approved
uses of valacyclovir include treatment of first or recurrent genital herpes,
suppression of frequently recurring genital herpes, as a 1-day treatment for
orolabial herpes, and as treatment for varicella and herpes zoster (Table
49–1). Once-daily dosing of valacyclovir for chronic suppression in persons
with recurrent genital herpes has been shown to markedly decrease the risk of
sexual transmission. In comparative trials with acyclovir for the treatment of
patients with zoster, rates of cutaneous healing were similar, but valacyclovir
was associated with a shorter dura-tion of zoster-associated pain. Higher doses
of valacyclovir (2 g four times daily) have also been shown to be effective in
prevent-ing CMV disease after organ transplantation when compared with placebo.
Valacyclovir is generally well tolerated, although nausea, headache, vomiting, or rash occasionally occur. At high doses, confusion, hallucinations, and seizures have been reported. AIDS patients who received high-dosage valacyclovir chronically (ie, 8 g/d) had an increased incidence of gastrointestinal intoler-ance as well as thrombotic thrombocytopenic purpura and hemolytic-uremic syndrome; this dose has also been associated with confusion and hallucinations in transplant patients. In a recent study, there was no evidence of increased birth defects in 181 infants who were exposed to valacyclovir during the first trimester.
FAMCICLOVIR
Famciclovir
is the diacetyl ester prodrug of 6-deoxypenciclovir, an acyclic guanosine
analog (Figure 49–2). After oral administration, famciclovir is rapidly
deacetylated and oxidized by first-pass metabo-lism to penciclovir. It is
active in vitro against HSV-1, HSV-2, VZV, EBV, and HBV. As with acyclovir,
activation by phosphorylation is catalyzed by the virus-specified thymidine
kinase in infected cells, followed by competitive inhibition of the viral DNA
polymerase to block DNA synthesis. Unlike acyclovir, however, penciclovir does
not cause chain termination. Penciclovir triphosphate has lower affinity for
the viral DNA polymerase than acyclovir triphosphate, but it achieves higher
intracellular concentrations. The most com-monly encountered clinical mutants
of HSV are thymidine kinase-deficient; these are cross-resistant to acyclovir
and famciclovir.
The
bioavailability of penciclovir from orally administered famciclovir is 70%. The
intracellular half-life of penciclovir triphosphate is prolonged, at 7–20
hours. Penciclovir is excreted primarily in the urine.
Oral famciclovir is
effective for the treatment of first and recur-rent genital herpes, for chronic
daily suppression of genital herpes, for treatment of herpes labialis, and for
the treatment of acute zoster (Table 49–1). One-day usage of famciclovir
significantly accelerates time to healing of recurrent genital herpes and of
herpes labialis. Comparison of famciclovir to valacyclovir for treatment of
herpes zoster in immunocompetent patients showed similar rates of cutaneous
healing and pain resolution; both agents shortened the duration of
zoster-associated pain compared with acyclovir.
Oral famciclovir is
generally well tolerated, although headache, nausea, or diarrhea may occur. As
with acyclovir, testicular toxicity has been demonstrated in animals receiving
repeated doses. However, men receiving daily famciclovir (250 mg every 12
hours) for 18 weeks had no changes in sperm morphology or motility. In a recent
study, there was no evidence of increased birth defects in 32 infants who were
exposed to famciclovir during the first trimes-ter. The incidence of mammary
adenocarcinoma was increased in female rats receiving famciclovir for 2 years.
PENCICLOVIR
The guanosine analog
penciclovir, the active metabolite of famci-clovir, is available for topical
use. Penciclovir cream (1%) shortens the duration of recurrent herpes labialis
or genitalis (Table 49–1). When applied within 1 hour of the onset of prodromal
symptoms and continued every 2 hours during waking hours for 4 days, median
time until healing was shortened by 17 hours compared with placebo. Adverse
effects are uncommon, although applica-tion site reactions occur in about 1% of
patients.
DOCOSANOL
Docosanol is a
saturated 22-carbon aliphatic alcohol that inhibits fusion between the plasma
membrane and the HSV envelope, thereby preventing viral entry into cells and
subsequent viralreplication. Topical docosanol 10% cream is available without a
prescription; application site reactions occur in approximately 2% of patients.
When applied within 12 hours of the onset of prodromal symptoms, five times
daily, median healing time was shortened by 18 hours compared with placebo in
recurrent orolabial herpes.
TRIFLURIDINE
Trifluridine
(trifluorothymidine) is a fluorinated pyrimidine nucleoside that inhibits viral
DNA synthesis in HSV-1, HSV-2, CMV, vaccinia, and some adenoviruses. It is
phosphorylated intra-cellularly by host cell enzymes, and then competes with
thymidine triphosphate for incorporation by the viral DNA polymerase (Figure
49–3). Incorporation of trifluridine triphosphate into both viral and host DNA
prevents its systemic use. Application of a 1% solution is effective in
treating keratoconjunctivitis and recurrent epithelial keratitis due to HSV-1
or HSV-2. Cutaneous application of trifluridine solution, alone or in
combination with interferon alfa, has been used successfully in the treatment
of acyclovir-resistant HSV infections.
INVESTIGATIONAL AGENTS
Valomaciclovir is an
inhibitor of the viral DNA polymerase; it is currently under clinical
evaluation for the treatment of patients with acute VZV infection (shingles)
and acute EBV infection (infectious mononucleosis).
Related Topics