Chapter: Biology of Disease: Infectious Diseases and Treatments
Treatment of Infectious Diseases
TREATMENT OF INFECTIOUS DISEASES
The treatment of infectious disease is almost entirely pharmacological although nursing care is obviously necessary in many cases and surgery may sometimes be used. The development and extensive uses of antibiotics and other drugs over the last 60 years in particular has had an enormous impact in reducing the number and effects of infectious diseases. Viruses and bacteria are the most important causes of infectious diseases in developed countries, while fungi, protozoa and helminths are of increased importance in the tropical climates of developing countries.
ANTIVIRAL DRUGS
Viruses are acellular and can only replicate by utilizing the metabolic processes of their host cell. It is difficult to target viruses inside host cells without damaging both infected and normal host cells hence there are relatively few effective antiviral agents. To be fully effective, antiviral drugs should ideally inhibit viral replication but not affect the reproduction of the host cell. Unfortunately, current antiviral drugs are not fully effective as all interfere to some extent with reproduction of the host cell and so produce adverse effects. Antiviral agents do not normally directly ‘kill’ the virus but, rather, inhibit their replication. Therefore, they must be administered for sufficient time to allow natural immune mechanisms to eradicate all the virions present. Thus antiviral therapy may well fail in severely immunocompromized patients.
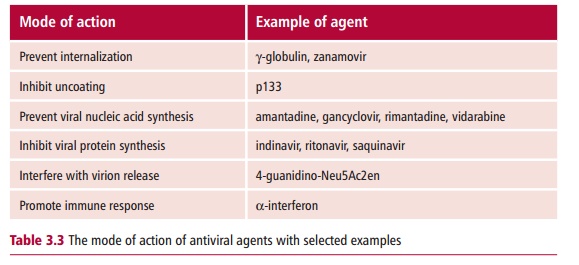
Most antiviral agents act by disrupting one of the steps in the replication cycle of the target virus. They may prevent viral internalization into the host cell. If the virion enters its host cell, other antiviral agents can interfere with the release of the viral nucleic acid from the capsid. Some antiviral agents prevent the synthesis of the viral nucleic acid or its proteins. Even if the virus is successfully synthesized, agents that interfere with its release from the host cell can prevent its dissemination. Lastly, some antivirals can help promote a more effective immune response against viral infection. Table 3.3 lists examples of each type of antiviral agents.

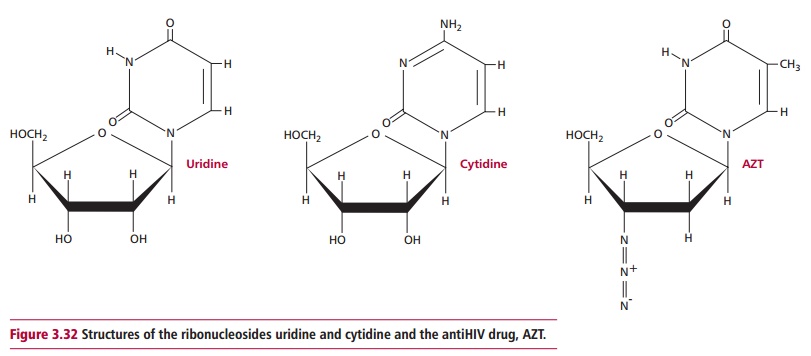
Viruses can become resistant to specific antiviral agents. Hence a therapy combining several agents, each of which acts at a different stage in the replication cycle is likely to be a more effective treatment. For example, HIV treatment is typically a combination of the antiHIV proteases (Figure 3.31) amprenavir, ritonavir and the antiHIV reverse transcriptase inhibitor AZT, which is a ribonucleoside analog (Figure 3.32).
ANTIBACTERIAL DRUGS
Drugs to treat bacterial diseases are generally called antibiotics. They are the most significant in clinical practice because bacteria are responsible for the greater proportion of infectious diseases. Most antibiotics are derived from the products of the metabolism of microorganisms, such as certain bacteria and fungi. Their actions usually rely on differences between microbial and host cells. Ideally, antibiotic drugs should kill the target bacterium, that is, they should be bactericidal. Bacteriostatic drugs prevent the bacteria replicating and must be administered for sufficient time to allow the immune defenses of the body to eliminate the pathogen.
Not all antibiotics are effective against all pathogenic bacteria. Some are only effective against Gram-negative bacteria, whereas others are effective against Gram-positive only (see later). Other antibiotics can be used to treat a range of both Gram-positive and negative organisms and are referred to as broadspectrum antibiotics. Antibacterial drugs generally act in one of four majorways: they may inhibit the synthesis of the bacterial nucleic acid, proteins or the cell wall or they may act in a variety of rather miscellaneous and, in some cases, unknown mechanisms.
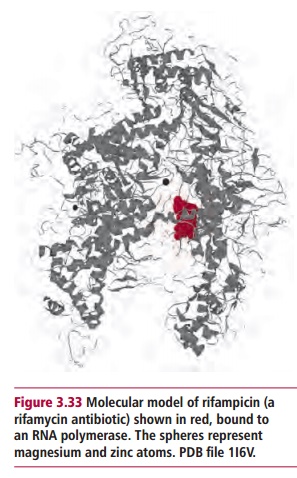
Many drugs inhibit both replication and transcription at several points. These drugs fall into a number of general families. Sulphonamides inhibit the formation of folate which is essential for the synthesis of precursors of nucleic acids. Nitroimidazoles bind directly to DNA and denature its helical structure such that it is no longer a substrate for DNA-binding proteins. Clofazimine, a drug which is used to treat leprosy, also binds to DNA preventing replication and transcription, though it is not a nitroimidazole. Quinolones, nalidixic acid and norfloxin, and the synthetic antibiotic ciprofloxacin, fluoroquinolones, offloxacin, norfloxacin and others, are inhibitors of DNA topoisomerase II, an enzyme essential for the replication and transcription of DNA. The rifamycins are inhibitors of RNA polymerases (Figure 3.33) and suppress transcription.
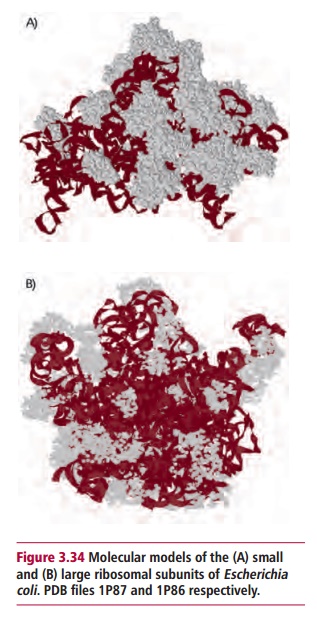
Protein synthesis begins with the translation of messenger RNA molecules by ribosomes to form polypeptides. Translation is broadly similar in both bacterial and mammalian cells though there are some significant differences between the two types of cells. For instance, bacterial ribosomes consist of 30S and 50S subunits (Figure 3.34), whereas eukaryotic ones have larger 60S and 40S subunits. A number of the protein translation factors necessary for translation also differ. These differences are exploited by a number of antibacterial drugs.
The main antibacterial antibiotics that interfere with protein synthesis are the aminoglycosides, lincosamides, macrolides and tetracyclines. The streptogramin quinupristin-dalfopristin, a relatively newly introduced drug, also interferes with protein synthesis. Aminoglycosides, such as streptomycin and gentamicin, are bactericidal and have complex effects following irreversible binding to specific proteins of the 30S subunit. They inhibit protein synthesis by interfering with initiation, inhibiting an essential checking step called proofreading performed by the ribosome so incorrect amino acids are inserted into the polypeptide leading to the production of nonfunctional or toxic peptides, inhibit elongation and prevent ribosomes associating together as functional polyribosomes. Clindamycin and lincomycin are lincosamides. These antibiotics can be bacteriostatic or bactericidal. They interfere with the first and subsequent steps in translation within the ribosome. Macrolides may be bacteriostatic or bactericidal. Examples include the widely used erythromycin and azithromycin and clarithromycin. They prevent translocation, that is, the movement of the peptide-tRNA complex within the ribosome. The tetracyclines are a well known group of drugs and include the parent tetracycline itself, as well as other antibiotics, such as doxycycline and oxytetracycline. Their action is bacteriostatic in that they inhibit the binding of the aminoacyl-tRNA complex to the 30S subunit and so slow down translation. Streptogramins type A and B (dalfopristin and quinupristin respectively), produced by Streptomyces pristinaepiralis, are chemically modified to give the drugquinupristin-dalfopristin. Quinupristin and dalfopristin alone each show weak bacteriostatic activities, however, together their actions are synergistic since they both target different site/actions of the 50S ribosome of bacteria, inhibit protein synthesis and lead to the release of incomplete polypeptides. Dalfopristin binds directly within the peptidyl transferase center of the ribosome interfering with the binding of tRNA molecules and inhibiting elongation of the polypeptide, while quinupristin blocks the exit tunnel of the polypeptide from the ribosome. Quinupristin-dalfopristin was licensed for use in the UK and USA in the late 1990s for treating severe infections with Gram-positive organisms, including nosocomial pneumonia and infections related to the use of intravascular catheters. It is particularly useful for treating complicated skin infections with methicillin-resistant Staphylococcus aureus and Streptococcus pyogenes and life-threatening infections of vancomycin-resistant Enterococcus faecium. Quinupristin-dalfopristin has poor activity against Enterococcus faecalis compared with Enterococcus faecium. The latter is generally a less serious pathogen and other treatments are available, even though the former is the more prevalent clinically. However, strains of Enterococcus faecium resistant to quinupristin-dalfopristin are being found.
Several other antibiotics that interfere with protein synthesis are also in clinical use. Fusidic acid, a bactericidal agent that is used only in the treatment of gonorrhea, inhibits translocation. Chloramphenicol is a widely known antibiotic although its action is bacteriostatic. It inhibits the formation of peptide bonds by the ribosome. Lastly, spectinomycin, used in the treatment of penicillin-resistant staphylococcal infections, prevents translocation.
Human cells, like all animal cells, lack a cell wall. This means that metabolic processes in the bacterial cell concerned with wall synthesis are excellent targets for specific antibacterial agents. The bacterial cell wall forms a protective bag around each microbial cell that prevents osmotic lysis. The wall contains layers of peptidoglycan, each of which consists of rows of amino sugars linked together by short peptides. Gram-negative bacterial cell walls have only a single layer of peptidoglycan. In contrast, those of Gram-positive organisms may have as many as 40.
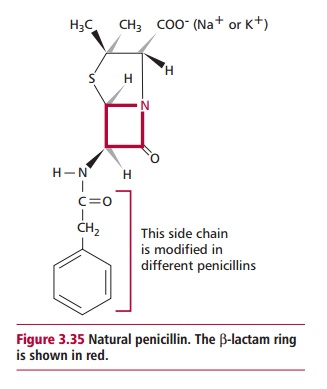
Bacterial growth involves cell division and entails the breakdown of the cell wall by bacterial enzymes, followed by synthesis of new peptidoglycan. Antibiotics, such as the A-lactams, that inhibit cell wall synthesis are almost all bactericidal since their presence stops new peptidoglycan formation and therefore affects cell wall synthesis while bacterial enzymes continue to break down the existing cell wall. A-Lactams are the largest, most widely used class of antibacterial antibiotics and contain the best known antibiotics, the penicillins and cephalosporins. They are, of course, only active against growing bacterial cells. All contain a chemical structure known as aA-lactam ring (Figure3.35) responsible for their antibacterial effects.A-Lactams are irreversibleinhibitors of transpeptidase, the enzyme that catalyzes the cross-link between the sugar residues and peptides in the peptidoglycan layer(s). A number of non-A-lactam antibiotics also reduce the efficiency of bacterial cell wall synthesis. They inhibit a number of different enzyme catalyzed steps in the synthesis at a wide variety of sites; hence they have no common mechanism of action. Examples of these drugs include cycloserine, vancomycin, fosfomycin and isoniazid among others.
A rather miscellaneous grouping of antibiotics includes the polymyxins, nitrofurantoin, pyrazinamide and metronidazole. Polymyxins are effective against Gram-negative bacteria where they disrupt the structure of the cell membrane. Nitrofurantoin may be considered a prodrug given the need for bacterial enzymes to metabolize it to an active form that is thought to damage bacterial DNA. Pyrazinamide is used to treat TB and acts by an unknown mechanism although TB treatment, in general, requires prolonged therapy with a combination of antibiotics (see later). Metronidazole is an effective drug against anaerobic bacteria and some protozoan parasites. The unionized form of metronidazole is readily taken up by these organisms, which possess electron transport systems able to reduce it to an active form that disrupts the helical structure of DNA, inhibiting bacterial nucleic acid synthesis. Metronidazole is used alone or combined with other antibiotics to treat abscesses in the liver, pelvis, abdomen and brain caused by susceptible anaerobic bacteria and colon infections caused by Clostridium difficile, Giardia infections of the small intestine, amebic liver abscesses and amebicdysentery and Trichomonas vaginalis vaginal infections, and both male and female carriers of trichomonas.
Despite their specificity, antibiotics can cause toxicity in humans including allergic responses to the penicillins and sulphonamides, ear and kidney damage by aminoglycosides, while chloramphenicol can show liver and bone marrow toxicities causing serious hematological diseases, particularly aplastic anemia. This can cause the death of rare susceptible individuals.
ANTIFUNGAL, ANTIPROTOZOAL AND ANTHELMINTHIC DRUGS
Fungi, protozoa and helminth parasites are responsible for many infections, particularly in the developing world. Given that they are all eukaryotic, then drugs to treat them are prone to act against host cells and they often have side effects. These drugs may kill the parasite or simply inhibit its growth. In the latter case, therapy must be continued for sufficient time to allow the host immune system to eradicate the organism.
Most antifungal drugs are not fungistatic but are fungicidal. One fungistatic drug, griseofulvin, inhibits intracellular transport and mitosis in fungi by interfering with the functions of their microtubules. A comparatively large number of fungicidal drugs suppress the synthesis of the essential cell membrane constituent, ergosterol. These include the allylamine antifungals, terbinafine and naftifine; the imidazoles, clotrimazole, econazole, ketoconazole and miconazole and the triazoles fluconazole and itraconazole. Ciclopiroxolamine inhibits the synthesis of fungal cell membrane proteins. The polyene antifungals, amphotericin and nystatin insert into plasma membranes of susceptible fungi. This increases the permeability of the membranes, allowing water and ions to leak and kill the parasite. Fluorocytosine inhibits the synthesis of fungal DNA.
In many cases, the precise biochemical mechanisms of antiprotozoal drugs are not known in any great detail. However, atovaquone inhibits electron transport in mitochondria. Pentamidine and isethionate interfere with the synthesis of protozoal macromolecules, while metronidazole, nifurtimox and tinidazole are thought to denature existing macromolecules. A number of other antiprotozoal therapeutic drugs are in clinical use. These variously affect protozoal enzymes, inhibit glycolysis and fatty acid oxidation or inhibit the synthesis of precursors of nucleic acids.
In general, the therapeutic bases of anthelminthic drugs are poorly understood. A number of commonly used drugs interfere with muscle contractions in the worms producing flaccid or spastic paralysis. This kills the parasite or makes it susceptible to attack by the host immune system. Paralysis is achieved by several overlapping mechanisms. Metriphonate inhibits cholinesterase leading to spastic paralysis while ivermectin potentiates inhibitory F aminobutyric acid mediated peripheral neurotransmission and levamisole and pyrantel block nerve transmission at the neuromuscular junction. Diethylcarbamazine and piperazine cause hyperpolarization of muscle membranes. Praziquantel acts directly on muscle cells and increases the permeability of muscle membranes to Ca2+.
Other anthelminthic agents act through different mechanisms. Oxamniquine interacts with helminth DNA and disrupts its structure. Niclosamide inhibits mitochondrial oxidative phosphorylation in helminth parasites. The anthelminthic agents albendazole, mebendazole and thiabendazole disrupt the microtubules of the cytoskeleton of the helminth.
COMBINATION THERAPY
Combination therapy is the treatment of infections with two or more drugs usually to increase the clinical efficacy of the treatment, for example as described above for quinupristin-dalfopristin, or to minimize the development of resistant strains of the infective organism. Where the infection is of unknown origin, then multiple therapies are advisable to fight the most likely pathogens. With mixed infections involving two or more known pathogens, it is desirable to target each microorganism with one or more different antibiotics. Combination therapy may be used even if only a single infective pathogen is present as using combinations of drugs can prevent or delay the development of resistance to the drugs being used. For example, some bacteria are resistant toA-lactams because theyproduce aA-lactamase that catalyzes the breakdown of the A-lactam ring. Combining an inhibitor of A-lactamase, such as clavulanic acid, with the A-lactam antibiotic helps preserve the drug in vivo. Other drugs commonly combined in therapeutic use are sulphamethoxazole and trimethoprim that synergistically inhibit the synthesis of folate by blocking different steps in its synthesis. The cocktail of isoniazid, rifampicin and pyrazinamide is used in the treatment of TB, while clofazimine, dapsone and rifampicin are used in combination in the therapy of leprosy.
SURGERY
Most infectious diseases can be treated using drug therapies. However, surgical intervention may be required in instances when the pathogen is resistant to available treatments or where it is the only means to contain an infection that is spreading to other areas of the body, as, for example, in gangrene caused by Clostridium perfringens(Figure 3.38) or necrotizing fasciitis (Margin Note3.6). In other cases, surgery might be desirable because access to the affectedsite by the antimicrobial agents is limited, as in the case of some abscesses or appendectomy where surgical drainage or removal of necrotic tissue respectively can enhance the recovery process.


Related Topics