Chapter: Biology of Disease: Infectious Diseases and Treatments
Investigating Infectious Diseases
INVESTIGATING INFECTIOUS DISEASES
To diagnose an infectious disease, two criteria must be satisfied. First, signs and symptoms compatible with the suspected infectious agent must be apparent. Secondly, the pathogen must be recovered from the infected site of the patient or there must be evidence of the pathogen being present at that site.
Infectious diseases can affect any organ or system and can cause a wide variety of symptoms and signs . The clinical history and examination should aim to identify the sites of infection and the causative organism. The clinical history focuses on aspects relevant to infectious disease, such as recent travel history, food and water intake, occupational exposure, sexual activity and any use of intravenous drugs. The clinical examination involves identifying fever, skin rashes, swollen lymph nodes (lymphadenopathy) and investigations of the eyes, ears, mouth and throat. Fever is a typical symptom of infection but not all patients with fever have an infection and not all infectious diseases present with fever. Investigations of the vagina, rectum and penis are necessary in sexually transmitted diseases.
The history and clinical examination is often supported by tests to assess health and identify the organs affected. These tests include imaging techniques such as X-rays, ultrasound, computerized tomography (CT), magnetic resonance imaging and blood tests. The blood tests involve a full blood count where eosinophilia is an important finding in parasitic infections and lymphocytosis is usually found in viral infections. The erythrocyte sedimentation rate and C-reactive protein levels are nonspecific tests of value in monitoring the course of infectious diseases. Other tests involve assessing liver and renal functions, which may be disrupted by an infection.
Microbiological tests to identify the infectious agents are especially helpful. Specimens for microbiological investigations include blood, cerebrospinal fluid (CSF), feces, pus, sputum and urine . Microbiological investigations use a variety of techniques, including culture, serological, biochemical and molecular biological tests.
CULTURE
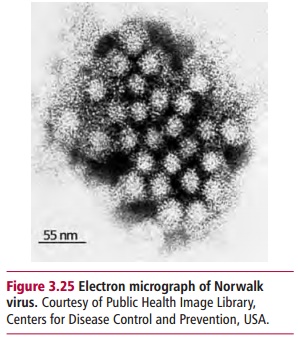
The suspected microorganism from the patient is grown outside the body usually on or in growth media, such as nutrient agar or broth, or in selective media which support the growth of particular microorganisms, until growth is detectable. In the case of bacteria, characterization is based on their microscopic appearance and the shape, texture and color of colonies. To some extent, bacteria can be identified by their ability to grow in specific media, such as blood agar or Sabouraud’s agar. Indicator media that include some substance that visibly changes as a result of the metabolic activities of particular microorganisms are also of use in identification. Fungi and mycoplasmas are cultured in similar ways to bacteria. However, they require a greater use of microscopic and colonial morphology for identification. Certain microorganisms, such as chlamydiae, and all human viruses, are intracellular pathogens and their growth requires the inoculation of cultured eukaryotic cells. When viruses infect and replicate within cultured cells, pathological changes are produced that are characteristic of particular viruses and can be used for identification purposes. Moreover, electron microscopy of supernatants from these cultures, or even on patient samples, can show the presence of particular viruses. For example, Norwalk virus (Figure 3.25), which causes outbreaks of vomiting and diarrhea, has been identified in stool samples of affected individuals. The diagnosis of parasitic infections, protozoa and helminths, may involve growing them in culture. More frequently, parasites are identified directly from specimens isolated from infected patients and/or indirectly by examining the cysts or eggs of the parasite
SEROLOGICAL TESTS
Serological tests involve identifying infectious agents indirectly by measuring serum antibodies in the affected individual. Antibody levels against a pathogen increase in the early stages of the disease and then fall during recovery. Such tests are particularly useful in situations where it is impossible to isolate the infectious agent and are used to make a diagnosis of, say, HIV infection. It is preferable to take a blood sample early in the infection (acute serum) and 10–14 days later (convalescent serum). A fourfold or greater rise in antibody titer in the second serum is diagnostic of an acute infection. Antibodies to bacteria can be detected by their ability to agglutinate these microorganisms. Alternatively, a variety of immunotechniques are available which are outlined.
BIOCHEMICAL AND MOLECULAR BIOLOGICAL TECHNIQUES
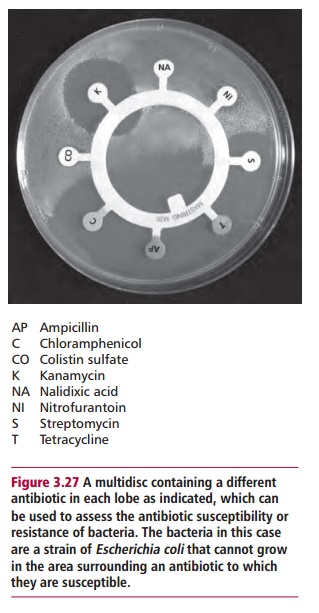
A variety of biochemical and molecular biological tests are available to help identify microorganisms. The simple Gram stain can immediately eliminate many possible bacteria. The abilities of some bacteria specifically to ferment some carbohydrates, use different substrates, express restricted enzyme activities or form specific products in enzyme catalyzed reactions can all aid in identification. The susceptibility of bacteria to different antibiotics (Figure 3.27) can also be useful and has the added utility of indicating possible therapy.
The GC ratio of the DNA of bacteria is generally expressed as a percentage 100(G + C) / (A + T + G + C) and varies from 20 to nearly 80%. Any one particular group of bacteria has a characteristic value. Specific genes from pathogenic organisms have been cloned and sequenced. That for the 16S ribosomal RNA has received considerable attention and, indeed, differences in the sequences of this gene have allowed evolutionary relationships between different groups of bacteria to be deduced. The entire genomes of a number of viruses, pathogenic bacteria and parasitic protozoa have been sequenced and the number is growing rapidly. Nucleic acid probes can be designed to detect characteristic sequences of pathogens and identify the organism in body fluids or tissues. This technique has been enhanced by the development of thepolymerase chain reaction.
The polymerase chain reaction
The polymerase chain reaction (PCR) was devised by Mullis in 1983. He was awarded the Nobel Prize in Chemistry in 1993; the shortness of time between the two dates is indicative of the perceived importance of PCR. Indeed, it is hard to exaggerate the impact of PCR because it has revolutionized molecular biological techniques. The PCR is an elegantly simple in vitro method for increasing in an exponential manner the number of relatively short strands of specific DNA fragments, normally of 500 to 5kbp, although longer lengths of up to 40 kbp can also be amplified. These may be whole genes but are more usually only a fragment of one. DNA consists of two polynucleotide strands arranged together in the now familiar double helical structure. The strands run in opposite directions: one in the 3'->5’ direction, the other with a 5’ -> 3’ orientation. The bases of the nucleotides of each strand associate together such that they form complementary pairs, with an adenine (A) of one strand paired with a thymine (T) of the other, and guanine (G) pairing with cytosine (C). The base pairs, and therefore the polynucleotide strands, are held by hydrogen bonds. It is the sequence of bases in the DNA strand(s) that is unique and specific to a particular gene.
The PCR is used to replicate the original DNA sample (template DNA) using a DNA dependent DNA polymerase, usually abbreviated to DNA polymerase. This enzyme copies the template DNA to produce new strands with complementary sequence. Some DNA polymerases can proofread, that is correct any mistakes in the newly formed strand to ensure the fidelity of its sequence. Crucially, DNA polymerases cannot begin the new strand de novo, but can only extend an existing piece of DNA. Thus two primer molecules are required to initiate the copying process. The primers are artificial oligonucleotides, short DNA strands of fewer than 50 nucleotides that are complementary to regions that flank the section of the template DNA of interest. Hence the primer determines the beginning of the region to be amplified. Primers are usually made to order by commercial suppliers who must, of course, be supplied with the required sequence.
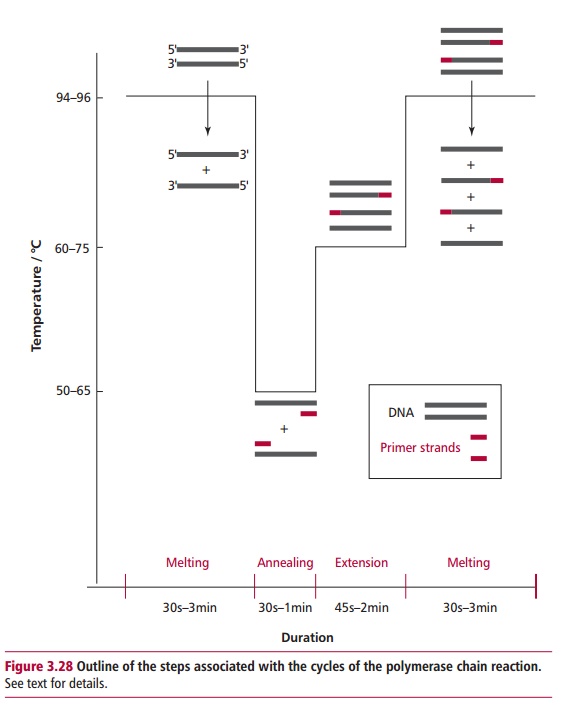
The PCR consists of a series of cycles, each of which consists of three identical steps (Figure 3.28). First, the double-stranded DNA has to be heated to 90– 96°C to break the hydrogen bonds that connect the two strands and allow them to separate. This denaturation is called melting. Prior to the first cycle, the DNA is often heated for an extended time, called a ‘hot start’, of up to 5 to 10 min to ensure that the template DNA and the primers are fully melted. In subsequent cycles, 30 s to 3 min at 94 to 96°C is normally sufficient for melting. Placing a heated lid on top of the reaction tubes or a layer of oil on the surfaces of the reaction mixtures prevents evaporation. The second step is primer annealing at temperatures of 50 to 65°C for 30 to 60 s, during which the primers bind to their complementary bases on the now single-stranded DNA templates. The primers must be present in excess of the target DNA otherwise its strands will simply rejoin. The design of the length of the primers requires careful consideration. Primer melting temperature, which must not be confused with that of the template DNA itself, increases with the length of the primer. The optimum length for a primer is generally 20 to 40 nucleotides that have melting temperatures of 60 to 75°C, although this depends upon their G/C content. If the primers are too short they will anneal at random positions on the relatively long template and result in nonspecific amplification. However, too long a primer and the melting temperature would be so high, that is above 80°C, that the DNA polymerase would have reduced
activity. The third step is the synthesis of DNA in reactions catalyzed by the polymerase and which extend the primers using the complementary strands as templates. This usually requires 45 to 120 s at 72°C. The extended portion of DNA is a complement of the template strand. Given the high temperatures of the reactions, DNA polymerases from thermophilic organisms are preferred. The Taq polymerase from Thermus aquaticus is widely used although it has the disadvantage of lacking proofreading capabilities and therefore of introducing errors (mutations) of 1 in 400 to 500 nucleotides in the new DNA. Polymerases such as Pwo or Pfu, obtained from Archea, have proofreading mechanisms that significantly reduce mutations and are used in ‘long range’ PCR of up to about 30 kbp.
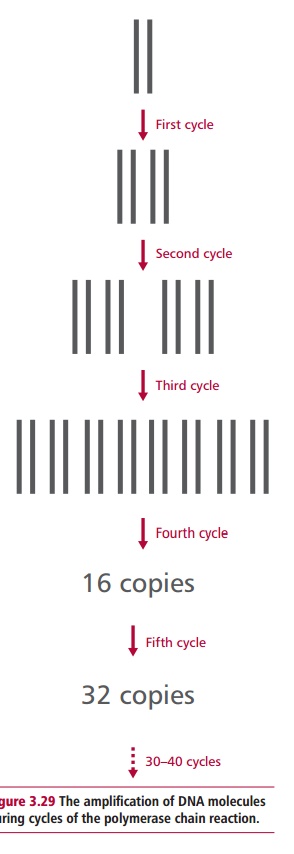
The result of the first cycle is two helices that are usually overextensions of the target sequences. Each is composed of one of the original strands plus its newly assembled complementary strand and its associated primer, for each of the original template DNA molecules. The cycle is usually repeated 20 to 30 times in identical conditions with a doubling of the amount of DNA present for every cycle (Figure 3.29). Hence after 30 cycles the amount of DNA in the original sample has increased over 230 (109 fold)! A PCR experiment normally terminates with a 10 min incubation at 72°C to ensure that all of the amplified DNA molecules are fully extended by the polymerase. Note that extremely stringent conditions are necessary to prevent any unwanted DNA contaminating PCR experiments, since this would also be amplified.
The great advantages of PCR are the increase in sensitivity and its automation. Samples of DNA from even a single cell or from samples many years old can be amplified and then analyzed. The PCR reaction is automated in a thermocycler, which automatically heats and cools the reaction tubes to the appropriate temperatures, for the desired times and the required numbers of cycles.
The products of PCR are identified by determining their base sequences and/ or their sizes using agarose or polyacrylamide gel electrophoresis against a known sample. The size of the product can be estimated by comparison with the electrophoretic mobility of fragments of DNA of known size (Figure 3.30).
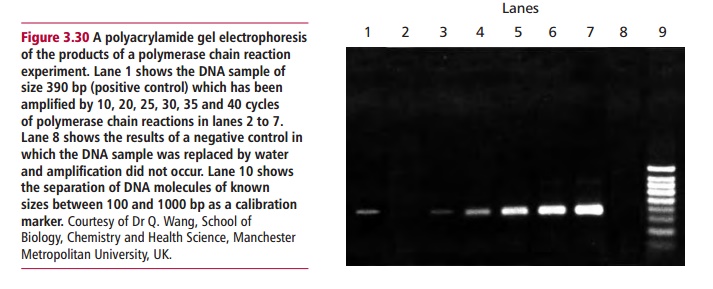
The biomedical sciences apply PCR in four main areas, which are the identification of infectious disease organisms for diagnostic purpose, in the detection of variations and mutations in hereditary diseases, detecting acquired mutations that lead to cancers and intissue typing . It is especially useful in diagnosing diseases caused by organisms that are difficult or impossible to culture. Its ability to amplify incredibly small amounts of DNA means that PCR-based tests can identify sources of infection more accurately, reliably, rapidly and cheaply than previous methods. For example, it can detect three different sexually transmitted
Polymerase chain reaction tests can even distinguish the specific strains of papillomaviruses that predispose individuals to cervical cancer. Diagnostic tests are also available for the viruses involved in AIDS, viral hepatitis and viral meningitis. The PCR is the most sensitive and specific test for Helicobacterpylori, the main cause of stomach ulcers . In England and Wales,47% of cases of meningococcal infections in 2002 were diagnosed using PCR tests for meningococcal DNA in clinical samples. Bacterial infections in middle ear fluid from children suffering otitis media are detectable by PCR, indicating an active infection, even when standard culture methods fail. The Lyme disease bacterium, Borrelia burgdorferi, is often difficult to diagnose accurately on the basis of general symptoms but PCR can amplify its DNA in body fluids.
Related Topics