Chapter: Biotechnology: Recombinant DNA Technology
Tools of rDNA technology
Tools of rDNA technology
In order to generate recombinant DNA molecules, we not only require the vector and insert DNA but also a method to precisely cut these DNA molecules and then join them together (ligation). Several molecular tools are required to perform the various steps and these are described in the following sections.
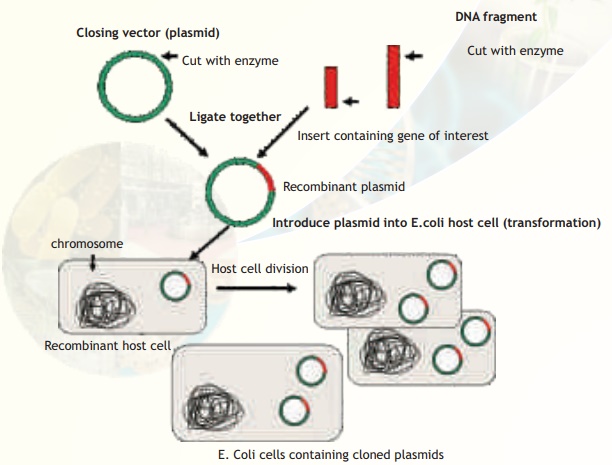
Restriction Enzymes: The Molecular Scissors
The foundations of rDNA technology were laid by the discovery of restriction enzymes. These enzymes exist in many bacteria where they function as a part of a defence mechanism called the Restriction-Modification System. This System consists of two components:
1. A restriction enzyme that selectively recognises a specific DNA sequence and digests any DNA fragment containing that sequence. The term restriction is derived from the ability of these enzymes to restrict the propagation of foreign DNA (e.g. Viral/phage DNA) in a bacterium.
2. A modification enzyme that adds a methyl group to one or two bases within the sequence recognised by the enzyme. Once a base is modified by methylation, the sequence cannot be digested. It is thus obvious that the Restriction-Modification enzyme system within a given bacterium protects its DNA from digestion by methylation but can digest foreign DNA which is not protected by similar methylation.
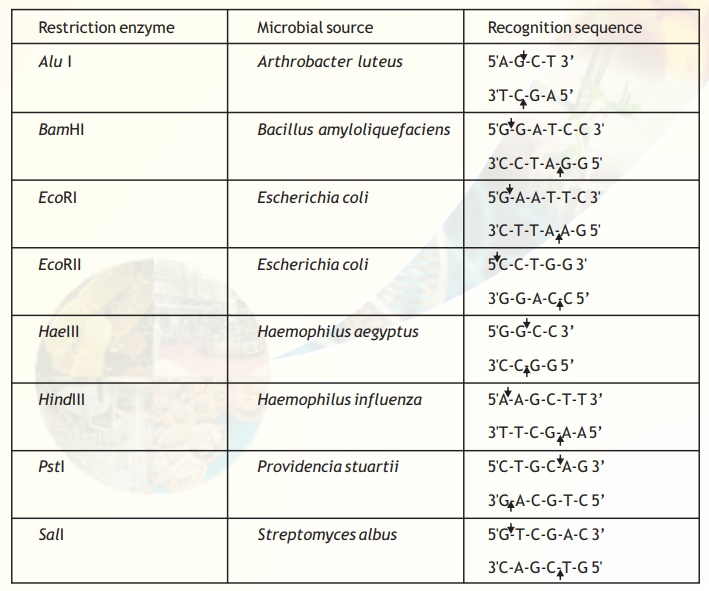
Different species of bacteria contain their own sets of restriction endonucleases and corresponding methylases. Three main classes of restriction endonucleases- type I, type II and type III are present, of which, only type II restriction enzymes are used in rDNA technology as they recognise and cut DNA within a specifc sequence typically consisting of 4-8 bp. This sequence is referred to as a restriction site and is generally palindromic, which means that the sequence in both DNA strands at this site read same 5' to 3' direction. Type II restriction enzymes are named after the bacterial species they have been isolated from. For example a commonly used restriction enzymeEcoRI isolated from the bacterial species E. coli is named so with the first three italicised alphabets referring to the genus (E) and species (co), the capital R referring to the strain (RY 13) and the number designated with the roman numeral (I) indicating that it was the first enzyme to be isolated from this strain of bacteria. Restriction enzymes were first discovered and studied by the molecular biologists W. Arber, H. Smith and D. Nathans for which they were awarded the Nobel Prize in 1978. Today more than a thousand restriction enzymes are available for genetic engineers to use. Some commonly used restriction enzymes (type II) along with their source and restriction- modification sequences have been listed in Table1 below.
Table 1. Type II restriction enzymes, their sources, recognition and cleavage sites.
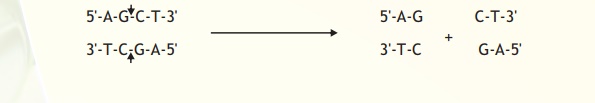
The exact kind of cleavage produced by a restriction enzyme is important in the design of a gene cloning experiment. Some cleave both strands of DNA through the centre resulting in a blunt or flush end. These are also known as symmetrical cuts. From Table I it is obvious that the enzyme AluI cuts symmetrically.
However EcoRI cuts in a way producing protruding and recessed ends known as sticky or cohesive ends because these ends can base pair and stick the DNA molecule back together again. Such cuts are termed staggered.
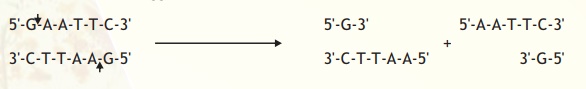
Note that EcoRI generates 5' overhangs at the cut site (and 3' recessed ends). Hence if two different DNA fragments containing EcoRI recognition sites are cleaved and mixed the sticky ends can bind and generate a hybrid or recombinant DNA (Fig.2)
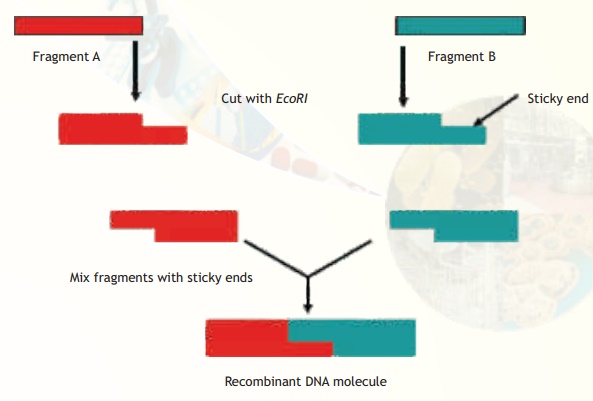
Fig. 2. Construction of rDNA using fragments from different sources.
Restriction Fragment Length Polymorphism (RFLP)
The DNA isolated from an individual organism has a unique sequence and even the members within a species differ in some parts of their sequence. The restriction sites would also vary and hence if DNA from a given individual was subjected to digestion with a restriction enzyme the fragments generated would vary when compared with another individual's DNA similarily digested. This variation in size (length) of the restriction enzyme generated fragments among individuals within a given species is termed RFLP. A schematic representation of how RFLPs are generated is given below (Fig. 3). A major application of this technique is DNA fingerprinting analysis which is explained below.
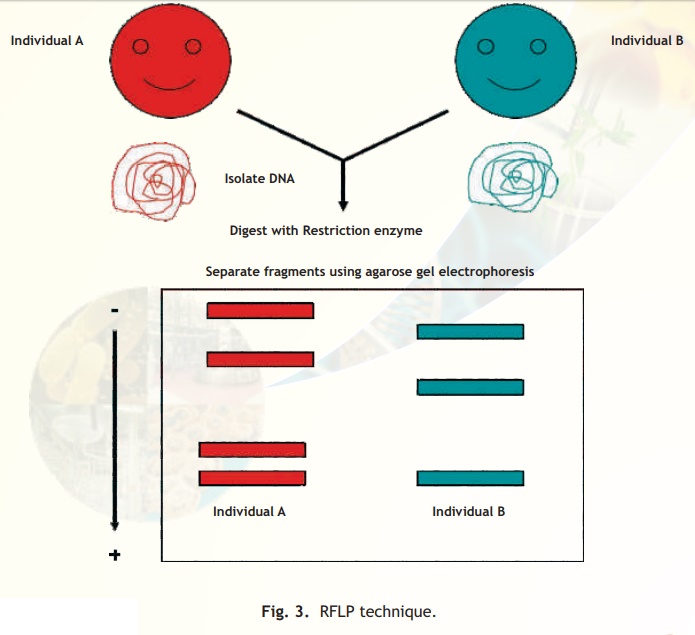
Fig. 3. RFLP technique.
Individuals except identical twins vary in their RFLP pattern as indicated schematically in the agarose gel electrophoresis. Hence the term DNA fingerprint is used and this is the basis of a major technique used in forensic science to identify and relate individuals.
Other enzymes used in cloning
In addition to restriction enzymes, there are several other enzymes that play an important role in rDNA technology. Two of these are DNA ligase and alkaline phosphatase.
DNA ligase: This enzyme forms phosphodiester bonds between adjacent nucleotides and covalently links two fragments of DNA . The reaction requires one of the fragments to have a 5' phosphate residue and the other a 3' hydroxyl group. In a previous section it was indicated how two fragments cut with EcoRI could stick together; DNA ligase seals this by forming a covalent bond. DNA ligase isolated from the bacteriophage T4 is frequently used to ligate different DNA fragments in order to generate rDNA molecules.
Alkaline phosphatase: Ligation requires the presence of a 5'phosphate group. If some of the fragments are treated with alkaline phosphatase to remove their phosphate groups then these cannot ligate within themselves and are forced to ligate with other fragments containing 5'phosphate groups. Hence this is a useful strategy to prevent self-ligation which would otherwise lead to wasteful ligation of fragments treated with restriction enzymes. An insert is ligated to the vector in generating rDNA as the vector is prevented from self-ligation by treating it with alkaline phosphatase. Alkaline phosphatase used for this purpose is purified from bacteria or calf intestines.
Vectors: Vehicles for cloning
Another major component of a gene cloning experiment is a vector such as a plasmid. A vector serves as a vehicle to carry a foreign DNA sequence into a host cell. A vector must possess certain features:
1. It must contain an origin of replication (ori) so that it is independently able to replicate within the host. This implies that any foreign insert it carries is automatically replicated.
2. It should incorporate a selectable marker, a gene whose product can identify the host cells containing the vector. Selectable markers include genes conferring antibiotic resistance, enzymes such as β-galactosidase which can turn substrates blue in the vicinity of the host cell colony and gene expressing Green Fluorescent Protein (GFP) which cause host cells containing the vector to fluoresce when viewed under UV light.
3. The vector must also have one unique restriction enzyme recognition site which can be used for cutting and introducing an insert. Most of the commonly used cloning vectors have more than one restriction site, they contain a Multiple Cloning Site (MCS) or polylinker. The MCS provides flexibility in the choice and use of restriction enzymes.
4. Another desirable feature of a cloning vector is that it should be small in size thereby facilitating entry/transfer into a host cell.
A number of vectors have been developed incorporating these features but only plasmids and bacteriophage based vectors will be discussed.
Plasmids
Plasmids are extrachromosomal, self-replicating, usually circular, double-stranded DNA molecules found naturally in many bacteria and also in some yeasts. Although plasmids are not essential for normal cell growth and division, they often confer useful properties to the host such as resistance to antibiotics that can be a selective advantage under certain conditions. The plasmid molecules can be present as 1 or 2 copies or in multiple copies (500-700) inside the host organism. These naturally occurring plasmids have been modified to serve as vectors in the laboratory and are by far the most widely used, versatile and easily manipulated vectors.
One of the earliest plasmid vectors to be constructed was pBR322 (Fig. 4a). This plasmid contains two different antibiotic resistance genes and recognition sites for several restriction enzymes. A popular series of plasmid cloning vectors is the pUC family (Fig. 4b). These vectors contain a region of the lacZ gene that codes for the enzyme β-galactosidase. This region also contains a polylinker and thus insertion of a foreign DNA into any of the restriction sites will result in an altered non-functional enzyme. During screening of recombinant plasmid containing host cells the absence of β-galactosidase activity is indicative of plasmids containing the insert.
The plasmid vectors described above can replicate only in E. coli. Many of the vectors used in eucaryotic cells are constructed such that they can exist both in the eukaryotic cell and E. coli. Such vectors are known as shuttle vectors. These vectors contain two types of origin of replication and selectable marker genes, one set which functions in the eukaryotic cells (e.g. yeast) and another in E. coli. An example of a shuttle vector is the yeast plasmid Yep (Fig. 4c). In the case of plants, a naturally occurring plasmid of the bacterium Agrobacterium tumefaciens called Ti plasmid has been suitably modified to function as a vector.
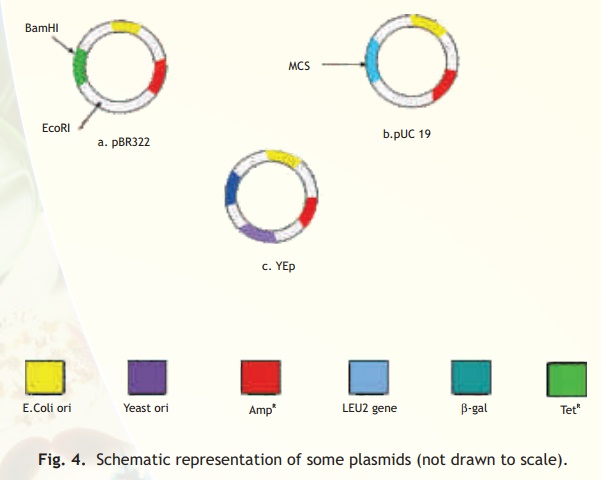
Fig. 4. Schematic representation of some plasmids (not drawn to scale).
The LEU2 gene (see Yep) codes for an enzyme which is needed for the synthesis of the amino acid leucine. Yeast cells having this plasmid can grow on a medium lacking leucine and hence can be selected over cells not containing the plasmid.
The discussion so far has focussed on cloning a DNA fragment to sufficiently amplify it. However the goal of the cloning experiment may be to produce a foreign protein in the host. In such cases the fragment containing the gene expressing the foreign protein is incorporated into the vector along with signals necessary for transcription and translation in the given host. Vectors which are suitable for expressing foreign protein are called expression vectors and one such vector is the pUC 19.
Vectors based on bacteriophages
Bacteriophages are viruses that infect bacterial cells by injecting their DNA into them and consequently take over the machinery of the bacterial cells to multiply themselves. The injected DNA hence is selectively replicated and expressed in the host bacterial cell resulting in a number of phages which eventually extrude out of the cell (lytic pathway) and infect neighbouring cells. This ability to transfer DNA from the phage genome to specific bacterial hosts during the process of viral infection gave scientists the idea that specifically designed phage based vectors would be useful tools for gene cloning experiments. Two phages that have been extensively modified for the development of cloning vectors are lambda (ë) and M13 phages.
Bacteriophage lambda has a double stranded, linear DNA genome containing 48,514 bp, in which 12 bases on each end are unpaired but complementary. These ends therefore are sticky and are called cohesive or cos sites and are important for packaging DNA into phage heads. An important feature of the lambda genome is that a large fragment in the central region of its genome is not essential for lytic infection of E. coli cells. Therefore, vectors have been designed such that this region can be replaced by foreign insert DNA. These phage based vectors allow cloning of DNA fragments up to 23 kb in size.
M13 is a filamentous phage which infects E. coli having a pilus (protrusion) which is selectively present in cells containing a F plasmid (called F+ cells). The genome of the M13 phage is a single stranded, circular DNA of 6407 bp. Foreign DNA can be inserted into it without disrupting any of the essential genes. In the life cycle of the phage following infection of the host E.coli cell the single stranded DNA is converted to a double-stranded molecule which is referred to as the Replicative Form (RF). The RF replicates until there are about 100 copies in the cell. At this point DNA replication becomes asymmetric and single stranded copies of the genome are produced and extruded from the cell packaged with protein as M13 particles (Fig. 5). The major advantages of developing vectors based on M13 are that its genome is less than 10 kb in size; the RF can be purified and manipulated exactly like a plasmid. In addition, genes cloned into M13 based vectors can be obtained in the form of single stranded DNA. Single stranded forms of cloned DNA are useful for use in various techniques including DNA sequencing and site-directed mutagenesis, techniques which will be discussed in a latter section.
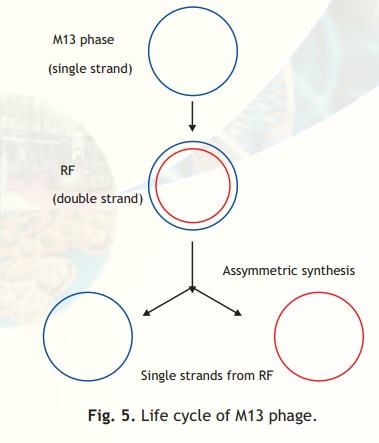
Fig. 5. Life cycle of M13 phage.
Cosmids
Cosmids have been constructed by combining certain features of plasmid and the 'cos' sites of phage lambda. The simplest cosmid vector contains a plasmid, origin of replication, a selectable marker, suitable restriction enzyme sites and the lambda cos site. Cosmids can be used to clone DNA fragments up to 45 kbp in size.
YAC vectors
YACs or Yeast Artificial Chromosomes are used as vectors to clone DNA fragments of more than 1 Mb in size. Therefore, they are useful in cloning larger DNA fragments as required in mapping genomes such as in the Human Genome Project. These vectors contain a teleomeric sequence, the centromere and an autonomously replicating sequence, features required to replicate linear chromosomes in yeast cells. These vectors also contain suitable restriction sites to clone foreign DNA as well as genes to be used as selectable markers.
BAC vectors
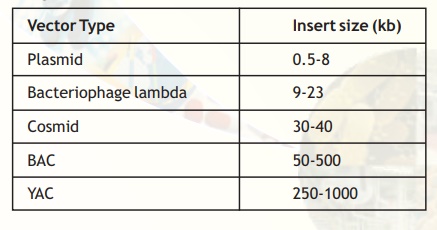
BACs or Bacterial Artificial Chromosomes are vectors based on the natural, extrachromosomal plasmid from E. coli - the fertility or F plasmid. A BAC vector contains genes for replication and maintenance of the F plasmid, a selectable marker and cloning sites. These vectors can accommodate inserts up to 500 kb and are used in genome sequencing projects. Table 2 lists the common cloning vectors with the size of insert that can be cloned into them.
Table 2. Common cloning vectors.
Animal and Plant viral vectors
You have already learnt how bacteriophages have been used to derive suitable vectors for gene cloning experiments in E. coli. Similarly, viruses that infect plant and animal cells have also been manipulated to introduce foreign genes into plant and animal cells. The natural ability of viruses to adsorb to cells, introduce their DNA and replicate, have made them ideal vehicles to transfer foreign DNA into eukaryotic cells in culture. A vector based on Simian Virus 40 (SV40) was used in the first cloning experiment involving mammalian cells. A number of vectors based on other type of viruses like Adenoviruses and Papillomavirus have been used to clone genes in mammals. At present, retroviral vectors are popular for cloning genes in mammalian cells. In case of plants, viruses like Cauliflower Mosaic Virus, Tobacco Mosaic Virus and Gemini viruses have been used with limited success.
Host Cells
The tools described in the previous sections will result in the generation of recombinant DNA molecules in the laboratory. Eventually the propagation of these DNA molecules must occur inside a living system or host. Many types of host cells including E. coli, yeast, animal and plant cells are available for gene cloning and the type of host cell used depends on the aim of the cloning experiment. E. coli has become the most widely used organism in rDNA technology because its genetic make-up has been intensively studied, it is easy to handle and grow, can accept a range of vectors and has been extensively studied for safety. Another major advantage of using E. coli as host cells is that under optimal conditions the cells divide every 20 minutes making it possible to clone large amounts of foreign DNA and if the appropriate signals are incorporated into the vector large amounts of recombinant proteins are available for therapeutics and other uses.
For the expression of eukaryotic proteins, eukaryotic cells are often preferred because to be functionally active, proteins require proper folding and post translational modifications such as glycosylation which is not possible in prokaryotic (E. coli) cells. Even cloned eukaryotic genes containing introns cannot be processed in E. colithereby necessitating the use of only eukaryotic host cells. Yeast cells have been used extensively for functional expression of eukaryotic genes because of several features. Yeasts are the simplest eukaryotic organisms (unicellular) and like E. coli have been extensively characterised genetically, easy to grow and manipulate and large amounts of cloned genes or recombinant proteins can be obtained from yeast cultures grown in fermentors (large culture vessels). Plant and animal cells may also be used as hosts in rDNA experiments and cells can be grown in tissue culture or can be induced and manipulated to form whole organisms (creation of transgenic animals and plants).
Related Topics