Chapter: Plant Biochemistry: A plant cell has three different genomes
The DNA of the nuclear genome is transcribed by three specialized RNA polymerases
The DNA of the nuclear genome is transcribed by three specialized RNA polymerases
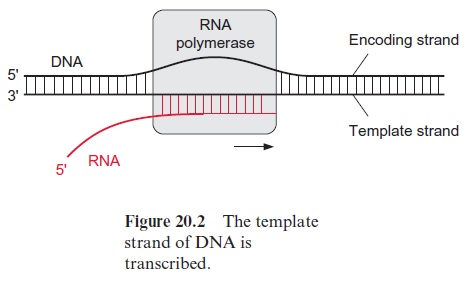
Of the two DNA strands, only the template strand is transcribed (Fig. 20.2). The DNA strand complementary to the template strand is called the coding strand. The latter has the same sequence as the transcription product RNA, with the exception that it contains thymine instead of uracil. The DNA of the nuclear genome is transcribed by three specialized RNA polymerases (I, II, and III)(Table 20.3). The division of labor between the three RNA polymerases, along with many details of the gene structure and principles of gene regulation, are valid for all eukaryotic cells. RNA polymerase II catalyzes the transcription of the structural genes and is strongly inhibited by α-amanitin at a concentration as low as 10–8 mol/L. α-Amanitin is the deadly poison from the toadstool Amanita phalloides (also called death cap). People frequently die from eating this toadstool.

The transcription of structural genes is regulated
In a plant containing about 25,000 to 50,000 structural genes, most of these genes are transcribed only in certain organs and then often only in certain cells. Moreover, many genes are only transcribed at specific times (e.g., the genes for the synthesis of phytoalexins after pathogenic infection). Therefore the transcription of most structural genes is subject to very complex and specific regulation. The genes for enzymes of metabolism or protein biosynthesis, which proceed in all cells, are transcribed more often. These genes, which every cell needs for such basic functions, independent of its specialization, are called housekeeping genes.
Promoter and regulatory sequences regulate the transcription of genes
Figure 20.3 shows the basic design of a structural gene. The section of the DNA on the left of the transcription starting point is termed 5’ or upstream and that to the right is referred to as 3’ or downstream. The encoding region of the gene is distributed among several exons, which are interrupted by introns.
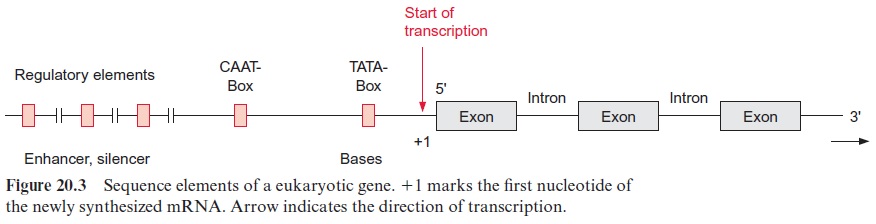
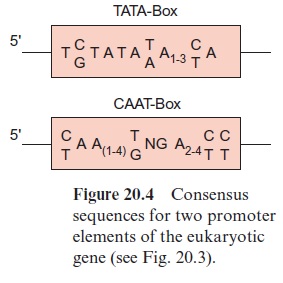
About 25 bp upstream from the transcription start site is situated a promoter element, which is the position where RNA polymerase II binds. The sequence of this promoter element can vary greatly between genes and between species, but it can be depicted as aconsensus sequence. (A consensus sequence is an idealized sequence in which each nucleotide is found in the majority of the sequences. Most of the promoter elements differ in their DNA sequences by only one or two nucleotides from this consensus sequence.) This consensus sequence is named the TATA box (Fig. 20.4). Another consensus sequence, the CAAT box, is often found about 80 to 110 bp upstream (Fig. 20.4). The housekeeping genes mentioned earlier often contain a C-rich region instead of the CAAT box. Additionally, sometimes more than 1,000 bp upstream, several sequences can be present, which function as enhancer or silencer(cis-regulatory elements).
Transcription factors regulate the transcription of a gene
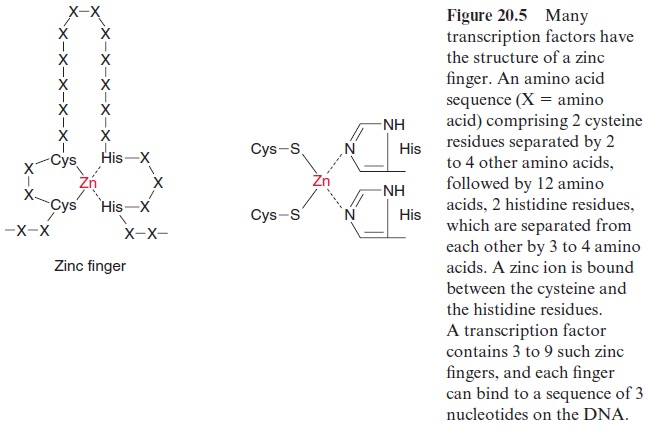
The regulatory elements contain binding sites for transcription factors (trans-factors), which are proteins modifying the rate of transcription. It has been estimated that the Arabidopsis genome encodes 1,500 factors for the regulation of gene expression. Different transcription factors often have certain structures in common. One type of transcription factor consists of a peptide chain comprising two cysteine residues and, separated from these by 12 amino acids, two histidine residues (Fig. 20.5). The two cysteine resi dues bind covalently to a zinc atom, which is also coordinatively bound to the imidazole rings of two histidine residues, thus forming a so-calledzinc finger. Such a finger binds to a nucleotide triplet of a DNA sequence. Zinc finger transcription factors usually possess several (up to nine) fingers and so are able to cling tightly to certain DNA sections.
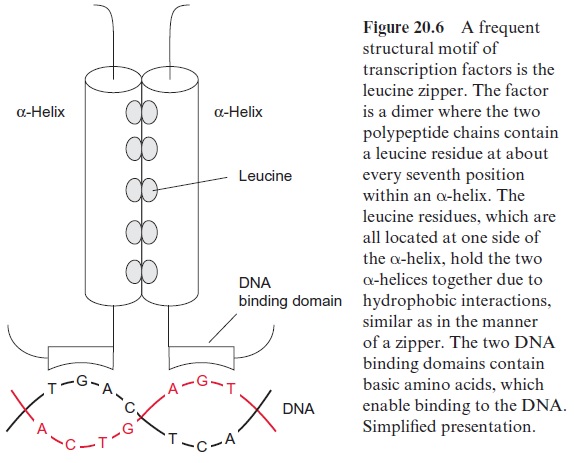
Another type of transcription factor is a dimer of DNA binding pro teins, where each monomer comprises a DNA binding domain and an -helix with three to nine leucine residues (Fig. 20.6). The hydrophobic leucine residues of the two α-helices are arranged in the dimer in such a way that they are exactly opposite each other like a zipper and both helices are held together by hydrophobic interaction. This typical structure of a transcription factor has been named the leucine zipper. The activity of transcrip tion factors is frequently regulated by signal chains linked to the perception of phytohormones or other stimuli.
Small (sm)RNAs inhibit gene expression by inactivating messenger RNAs
smRNAs, consisting of 21–24 nucleotides regulate the expression of genes that are involved in responses to stress and insufficient supply of nutritients.
Several smRNA types (miRNA, siRNA, nat-siRNA, and pre-miRNA) bind to complementary sequences of target mRNAs to form a double-strand RNA (dsRNA). This leads to the degradation of mRNA by RNAse II (Dicer) and other RNA degrading enzymes, which ultimately results in an inhibition of translation. In this way smRNAs function as negative regulators that interfere specifically with plant development. This property is utilized in biotechnology to suppress the expression of a certain gene by the so-called RNA interference (RNAi) technique. Plants can be made to synthesize a small RNA complemen tary to a defined mRNA by gene modification. The RNAi technique is also used to identify the function of a certain gene, by inhibiting its expression and evaluating the effects to the plant. In 2006 Andy Fire and Craig Mello (USA) were awarded the Nobel Prize for their basic studies of the RNAi technique.
The transcription of structural genes requires a complex transcription apparatus
RNA polymerase II consists of 8 to 14 subunits, but, on its own, it is unable to start transcription. Transcription factors are required to direct the enzyme to the start position of the gene. The TATA binding protein, which recognizes the TATA box and binds to it, has a central function in tran scription (Fig. 20.7). The interaction of the TATA binding protein with RNA polymerase requires a number of additional transcription factors (designated as A, B, F, E, and H in the figure). They are all essential for transcription and are termedbasal factors.
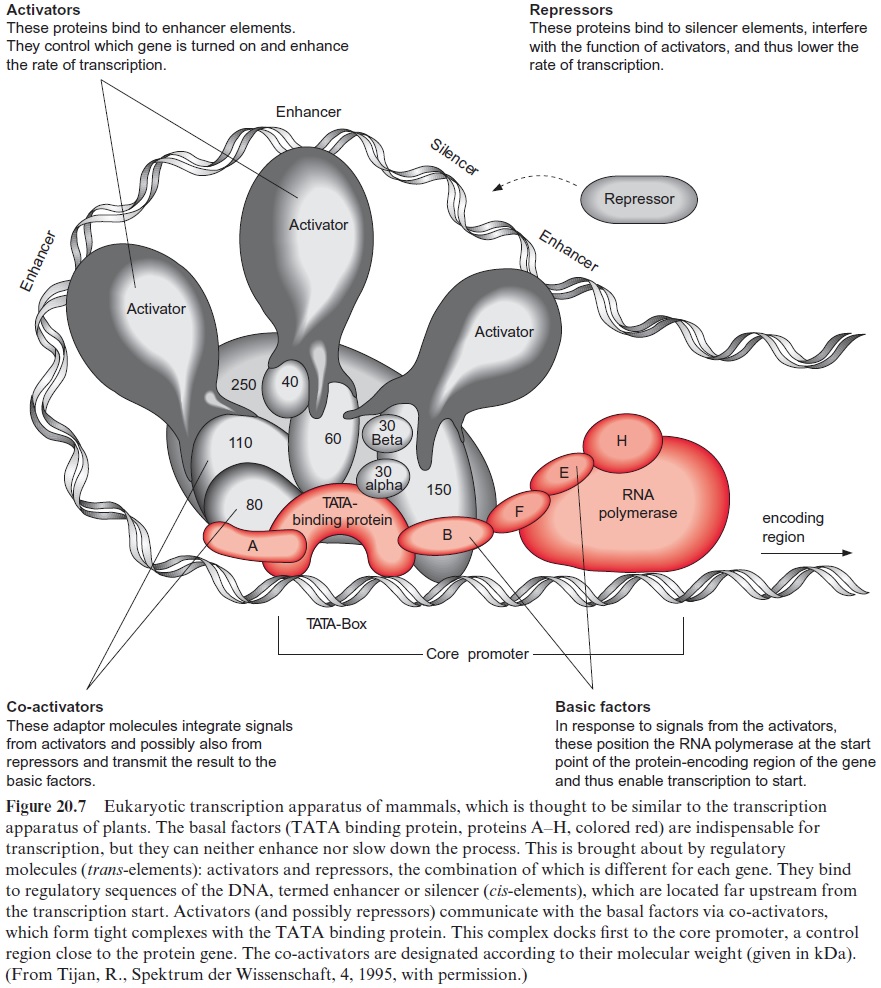
The transcription apparatus is a complex of many protein components, around which the DNA is wrapped in a loop. In this way cis-regulatory ele ments positioned far upstream or downstream from the encoding gene are able to influence the activity of RNA polymerase. The rate of transcription is determined by transcription factors, either activators or repressors (sum marized as trans-elements, since they are encoded by other regions of the DNAs) which bind to the upstream regulatory elements (enhancer, silencer;Fig. 20.3, called cis-elements, since they are part of the promoter of the encoding gene).
These transcription factors interact through a number of co-activators with the TATA binding protein and modulate its function on RNA polymerase. Various combinations of activators and repressors thus lead to activation or inactivation of gene transcription. The scheme of the tran scription apparatus shown in Figure 20.7 was derived from investigations with animals but it turned out that the regulation of transcription in plants resembles that of animals.
Knowledge of the promoter and enhancer/silencer sequences is very important for genetic engineering of plants . It is not always essential for this to know all these boxes and regulatory elements in detail. It can be sufficient for practical purposes if the DNA region is identified that is positioned upstream of the structural gene and influences its transcription in a specific way. In eukaryotic cells, this entire regulatory section is often sim ply called a promoter. For example, promoter sequences have been identified, which determine that a gene is to be transcribed in a leaf, and there only in the mesophyll cells or the stomata, or in potato tubers, and there only in the storage cells. In such cases, the specificity of gene expression is explained by the effect of cell-specific transcription factors on the corresponding promoters.
The formation of the mature messenger RNA requires processing
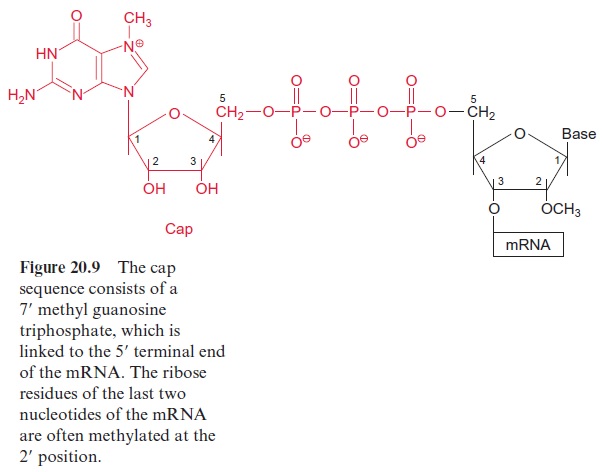
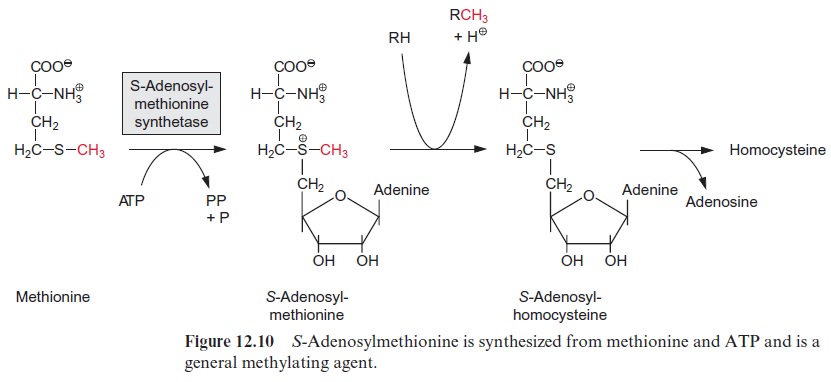
The transcription of DNA in the nucleus by RNA polymerase II yields a primary transcript (pre-mRNA, Fig. 20.8), which is processed in the nucleus to mature mRNA. During transcription, a GTP is hydrolyzed and a GDP molecule is linked to the 5’ -P group of the RNA terminus resulting in a triphosphate bridge (G capping) (Fig. 20.9). Moreover, guanosine and the second ribose (sometimes also the third, not shown in the figure) are methy lated using S-adenosylmethionine as a donor (Fig. 12.10). This modified GTP at the beginning of RNA is called the 5’ cap and is present only in eukaryotic mRNA. This cap functions as a binding site in the formation of the initiation complex during the start of protein biosynthesis at the ribo somes and probably also provides protection against degra dation by exo-ribonucleases.
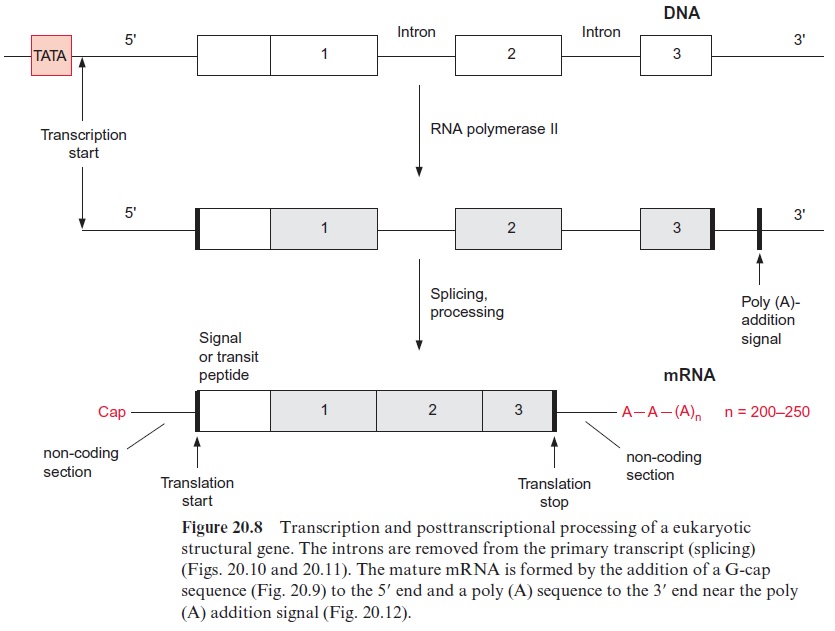
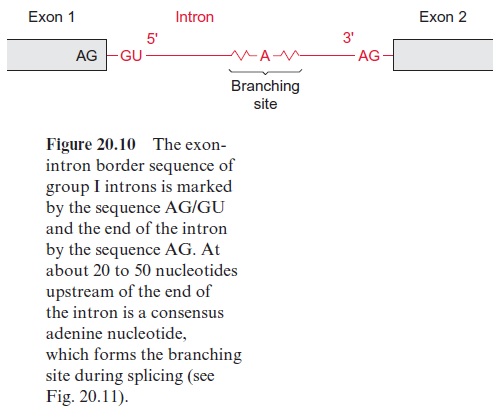
The introns are removed during further processing of the pre-mRNA by a process called splicing. The size of the introns can vary from 50 to over 10,000 nucleotides. Four types of introns are known. In the introns of group I the border sequences are highly conserved, the last two nucleo tides of the exon are in 95% of the cases AG and the first nucleotides of the intron are GU and the last are AG (Fig. 20.10). About 20 to 50 nucleo tides upstream from the 3 end of the intron an adenyl residue is known as the branching site. In the other intron types the border sequences are less conserved.
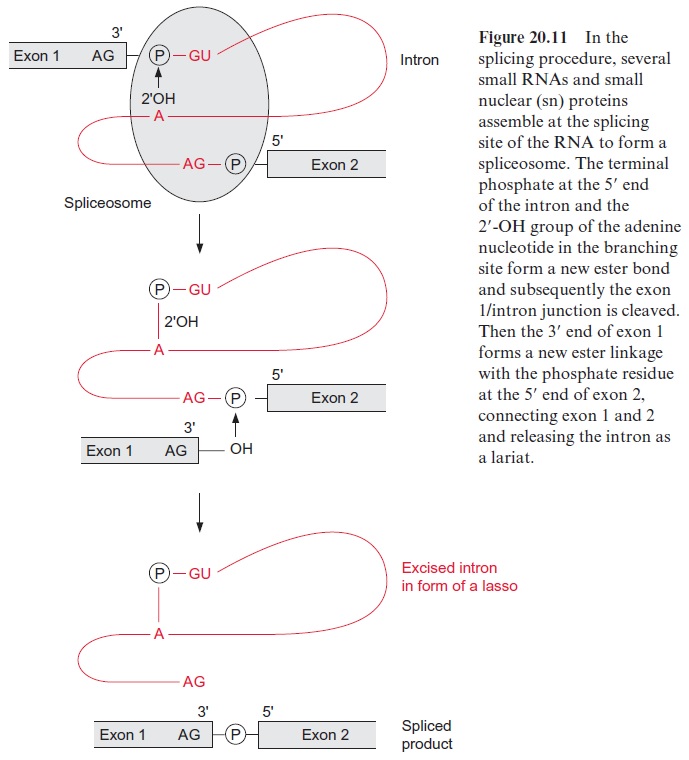
The excision (splicing) of group I introns is catalyzed by riboprotein com-plexes, composed of RNA and proteins. Five different RNAs of 100 to 190 nucleotides, named snRNA (sn=small nuclear), are involved in the splic ing procedure. Together with proteins and the RNA to be spliced, these snRNA form the spliceosome particle (Fig. 20.11). The first step is that the 2’ -OH group of the ribose of the nucleotide of the branching site forms a phosphate ester with the phosphate residue, linking the end of exon 1 with the start of the intron and cleaving the ester bond between exon 1 and the intron. This is followed by a second esterification between the 3’ -OH group of exon 1 and the phosphate residue at the 5’ -OH of exon 2, accompanied by a cleavage of the phosphate ester with the intron. Two transesterifica tion reactions complete the splicing process. The intron remains in the form of a lasso (lariat) and is later degraded by ribonucleases.
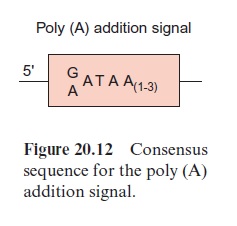
As a further step in RNA processing, the 3’ end of the pre-mRNA is cleaved behind a poly(A) addition signal (Fig. 20.12) by an endonuclease, and a poly(A)-sequence of up to 250 bp is added at the cleaving site. The resulting mature mRNA is bound to special proteins and leaves the nucleus as a DNA-protein complex.
rRNA and tRNA are synthesized by RNA polymerase I and III
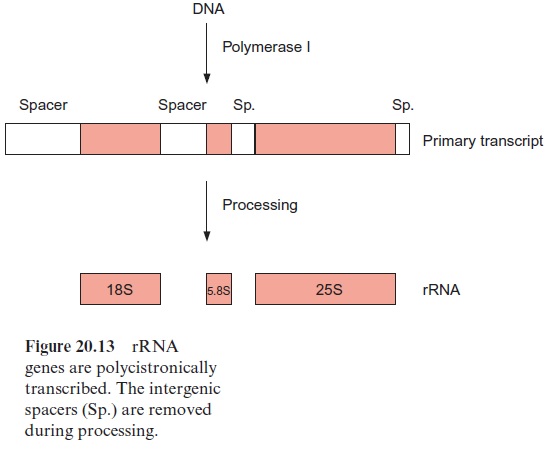
Eukaryotic ribosomes of plants are made of four different rRNA molecules named 5S-, 5.8S-, 18S-, and 25S-rRNA according to their sedimentation coefficients. The genes for 5S-rRNA are present in many copies, arranged in tandem on certain regions of the chromosomes. The transcription of these genes and also of tRNA genes is catalyzed by RNA polymerase III. The three remaining ribosomal RNAs are encoded by a continuous genome sequence, again in tandem and in many copies. These genes are transcribed byRNA polymerase I. The primary transcript is subsequently processed after methylation, especially of -OH groups of ribose residues, followed by the cleavage of RNA to produce mature 18S-, 5.8S-, and 25S-rRNA (Fig. 20.13). The excised RNA sequences between these rRNAs (intergenetic spacers) are subsequently degraded. Because of their rapid evolution, com parative sequence analyses of these spacer regions can be used to establish a phylogenetic classification of various plant species within a genus.
Related Topics