Chapter: Plant Biochemistry: A plant cell has three different genomes
DNA polymorphism yields genetic markers for plant breeding
DNA polymorphism yields genetic markers for plant breeding
An organism is defined by the DNA nucleotide sequences of its genome. Differences between the DNA sequences (DNA polymorphisms) exist only between different species, but also to some extent between individuals of the same species. Between two varieties of a cultivar, in a structural gene often 0.1% to 1% of the nucleotides are altered, mainly in the introns.
Plants usually are selected for breeding purposes by external features (e.g., for their yield or the resistance to certain pests). In the end, these characteristics are all due to differences in the nucleotide sequence of the genome. Selecting plants for breeding would be much easier if it were not necessary to wait until the phenotypes of the next generation were evident, but if instead the corresponding DNA sequences could be analyzed directly. A complete comparative analysis of these genes is not practical, since most of the genes involved in the expression of the phenotypes are not known, and the minute variation (0.1–1 %) of nucleotide sequence of these genes is only detectable with a large analytical expenditure. Other techniques that are easier and less expensive to carry out are available, as will be described in the following.
Individuals of the same species can be differentiated by restriction fragment length polymorphism
It is possible to detect differences in the genes of individuals within a species even without a detailed sequence comparison and to relate these differences empirically to analyzed properties. One of the methods for this is the analysis of restriction fragment length polymorphism (RFLP). It is based on the use of bacterial restriction endonucleases, which cleave a DNA at a palindromic recognition sequence, known as a restriction site (Fig. 20.14). The various restriction endonucleases have specific recognition sites of four to eight base pairs (bp). Since these restriction sites appear at random in DNA, those rec ognition sequences with 4 bp appear more frequently than those with 8 bp. Therefore it is possible to cleave the genomic DNA of a plant into thousands of defined DNA fragments by using a particular restriction endonuclease (usually enzymes with 6 bp restriction sites). The exchange of a single nucle otide in a DNA may eliminate or newly form a restriction site, resulting in a polymorphism of the restriction fragment length.

The digestion of genomic DNA with restriction enzymes results in a mul titude of fragments. In order to mark fragments of defined regions of the genome, labeled DNA probes are required. Such probes are prepared by the identification of a certain DNA region of a chromosome of about 10 to 20 kbp, located as near as possible to the gene responsible for the trait of interest, or which is even part of that gene. This DNA section is introduced into bacte ria (usually Escherichia coli) using plasmids or bacteriophages as a vector and is propagated there. The plasmids or bacteriophages are iso lated from the bacterial suspension; the multiplied DNA sequences are cut out again, isolated, and radioactively labeled or provided with a fluorescence label. Such probes used for this purpose are called RFLP markers.
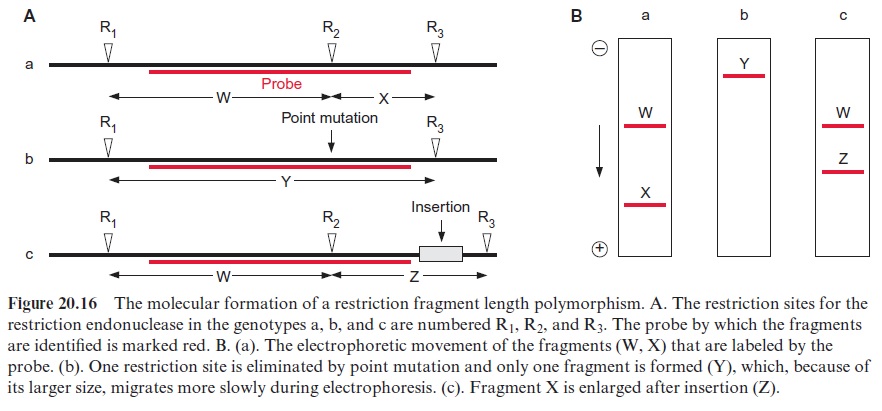
The analysis of DNA restriction fragments by the labeled probes is carried out by the Southern blot method, developed by Edwin Southern (Edinburgh) in 1975. The restriction fragments are first separated accord ing to their length by electrophoresis in an agarose gel (the shortest frag ment moves the farthest). The separated DNA fragments in the gel are transferred to a nitrocellulose or nylon membrane by placing the mem brane on the gel. By covering it with a stack of tissue paper, a buffer solu tion is drawn through the gel and the membrane, and the diffusive DNA fragments are bound to the membrane. The buffer also causes the dissocia tion of the DNA fragments into single strands (Fig. 20.15). When a labeled DNA probe is added, it hybridizes to complementary DNA sequences on the membrane. Only those DNA fragments that are complementary to the probe are labeled, and after removal of the nonbound DNA probe mol ecules by washing, are subsequently identified by autoradiography (in the case of a radioactive probe) or by fluorescence measurement. The position of the band on the blot is then related to its migration and hence its size.
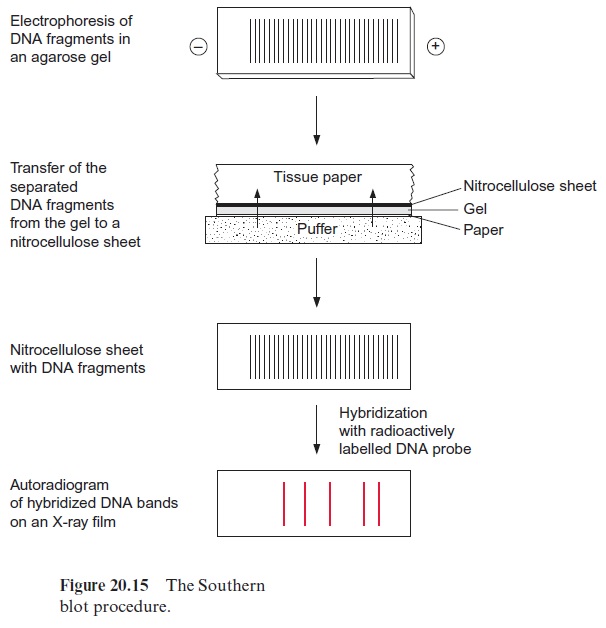
Figure 20.16 explains the principles of RFLP. Figure 20.16A shows in
(a) a gene with three restriction sites (R1, R2, and R3). Since the labeled probe binds only to the DNA region between R1 and R3, just two noticea ble restriction fragments (W and X) will be observed in the autoradiogram. Due to their different lengths, they are separated by gel electrophoresis and detected by hybridization with a probe (Fig. 20.16B (a)). Upon the exchange of one nucleotide (point mutation) (b), the restriction site R2 is eliminated and therefore only one labeled restriction fragment (Y) is detected, which, because of its larger size, migrates in gel electrophoresis slower than the fragments of (a). When a DNA section is inserted between the restriction sites R2 and R3 (c), the corresponding fragment (Z) is longer.
The RFLP represent genetic markers, which are inherited according to Mendelian laws and can be employed to characterize a certain variety. Normally several probes are used in parallel measurements. RFLP is also used in plant systematics to establish phylogenetic trees. Moreover, defined restriction fragments can be used as labeled probes to localize certain genes on the chromosomes. In this way chromosome maps have been established for several plants (e.g., Arabidopsis, potato, tomato, and maize).
The RAPD technique is a simple method for investigating DNA polymorphism
An alternative method for analyzing the differences between DNA sequences of individuals or varieties of a species is the amplification of ran domly obtained DNA fragments (random amplified polymorphic DNA, RAPD). This method, which has been in use only since 1990, is much easier to work with, compared to the RFLP technique, and its application has become widespread in a very short time.
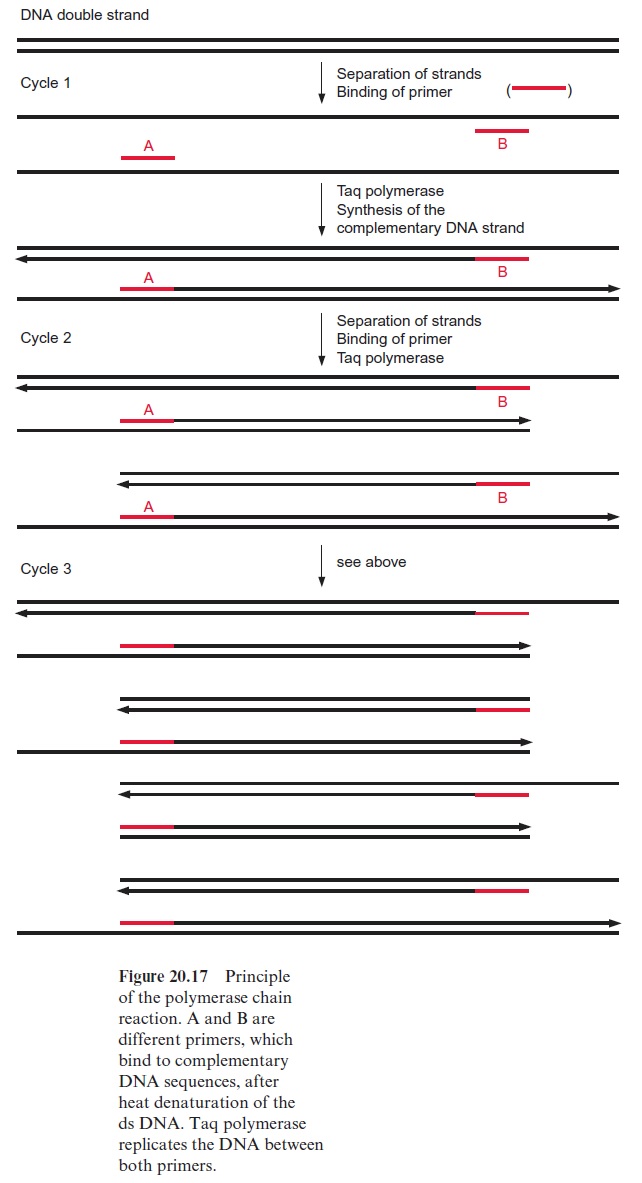
The basis for the RAPD technique is the polymerase chain reaction (PCR). The method enables selected DNA fragments of a length of up to two to three kbp to be amplified by DNA polymerase (Fig. 20.17). This requires an oligonucleotide primer (A), which binds to a complementary sequence of the DNA to be amplified by a special DNA polymerase and indicates the starting point for the synthesis of a DNA daughter strand at the template of the DNA mother strand. A second primer (B) is needed to define the end of the DNA strand that is to be amplified. In the first step, the DNA double strands are separated into single strands by heating to about 95°C. During a subsequent cooling period, the primers hybridize with the DNA single strands and thus enable, in a third step at a medium temperature, the synthe sis of DNA. A DNA polymerase originally isolated from the thermophilic bacteriumThermus aquaticus which lives in hot springs (Taq polymerase) is used, since this enzyme is not affected by the heat treatments. Subsequently, the DNA double strands thus formed are separated again by being heated at 95°C, the primer binds during an ensuing cooling period, and this is fol lowed by another cycle of DNA synthesis by Taq polymerase. The alternat ing heating and cooling can be continued for 30 to 40 cycles, and the amount of DNA is doubled during each cycle. It should be noted that during the first cycle, the length of the newly formed DNA is only defined at one end. During the second cycle the binding of the primer to the complementary nucleotide sequence of the newly formed DNA strand results in the synthesis of a DNA that is restricted in its length by both primers. With the increasing number of cycles, DNA fragments of uniform length are amplified. Since in the polymerase chain reaction the number of the DNA molecules formed is multiplied exponentially by the number of cycles (e.g., after 25 cycles by the factor 34×106) very small DNA samples (in the extreme case a single mol ecule) can be multiplied ad libitum.
In the RAPD technique, genomic DNA and only one oligonucleotide primer consisting typically of 10 nucleotides are required for the polymer ase chain reaction. Since the probability of the exact match of 10 comple mentary nucleotides on the genomic DNA is low, the primer binds at only a few sites of the genomic DNA. Characteristically the DNA polymerases used for this amplification require a distance between the two primers to be no larger than 2,000–3,000 bp. Therefore, only a few sections of the genome are amplified by the polymerase chain reaction and a subsequent selection of the fragments by a probe is not necessary. The amplification produces such high amounts of single DNA fragments that, after being separated by gel electrophoresis and stained with ethidium bromide, the fragments can be detected as fluorescent bands under ultraviolet (UV) light. Point muta tions, which eliminate primer binding sites or form new ones, and deletions or insertions, all of which affect the size and number of the PCR products, can change the pattern of the DNA fragments in analogy to the RFLP technique (Fig. 20.16). Changing the primer sequence can generate differ ent DNA fragments. Defined primers of 10 nucleotides are commercially available in many variations. In the RAPD technique, different primers are tried until, by chance, bands of DNA fragments that correlate with a certain trait are obtained. The RAPD technique takes less work than the RFLP technique because it requires neither the preparation of probes nor the time-consuming procedure of a Southern blot. It has the additional advantage that only very small amounts of DNA (e.g., the amount that can be isolated from the embryo of a plant) are required for analysis. Although in most cases it is not possible to define from which gene these fragments derive, the RAPD technique allows differentiation between varieties of a species, and has therefore become an important tool in breeding.
The polymorphism of micro-satellite DNA is used as a genetic marker
Recently micro-satellite DNA has become an important tool for identifying certain plant lines. Micro-satellite DNAs comprise sequences of one to two, sometimes also three to six, nucleotide pairs, which are located in 10 to 50 repetitions at certain sites of the genome, in the region of the intron, or directly before or behind a gene, whereby the number of the repetitions is highly polymorphic. Also, in this method PCR is utilized for detection. Not only is micro-satellite polymorphism used to identify individual humans (e.g., in criminal cases), but also is employed as a genetic marker for plant breeding.
Related Topics