Chapter: Biotechnology: Protein Structure And Engineering
Structure-Function relationship in Proteins
Structure-Function relationship in Proteins
As you have now learnt about how various forces drive proteins to assume characteristic shapes, it is worthwhile to consider why shape is paramount to the function of a protein. We will look at two proteins- an enzyme, chymotrypsin and the oxygen carrying protein, haemoglobin, to emphasise the importance of protein structure in its function.
Chymotrypsin, a proteolytic enzyme
As injested food makes way into the duodenum from the stomach, the proteins encounter a fierce proteolytic duo- trypsin and chymotrypsin which precisely cut the linear chains into short peptides which later on are acted upon by peptidases to release amino acids. Chymotrypsin, which hydrolyses peptide bonds following bulky aromatic amino acid residues in polypeptides is actually synthesised in the pancreas and through the pancreatic duct released into the duodenum. Have you wondered why this enzyme being a powerful proteolytic enzyme does not end up cutting cellular proteins within the pancreas itself? Nature has ensured that chymotrypsin and other proteolytic enzymes are synthesised as inactive harmless precursors known as zymogens which are then activated when required only in the duodenum, their site of activity, a process called in-situ activation. This activation in molecular terms results in an alteration in its shape so that it may now be able to interact with its substrate. The inactive precursor enzyme is termed chymotrypsinogen and the fully active enzyme is called chymotrypsin. The enzyme chymotrypsin is made up of a linear chain of 245 amino acids interrupted into three peptides-A,B,C. The protein folds into a globular structure. In the 3-D structure of the enzyme three important amino acid residues, his57, asp102 and ser195 come close together in space (Fig. 4) which allows a "charge relay system" to operate as indicated in Fig 5. The negatively charged asp102 is able to hydrogen bond with the adjacent his57 partially borrowing the hydrogen ion from the latter. The his57 makes good its partial hydrogen ion loss to aspartate by attracting a hydrogen ion from the adjacent ser195 through the his57 residue much like a relay race where the baton is passed from one member to another, the difference here being that the baton is a charge.
Normally the hydroxyl group of a serine residue is not acidic (pKa 12) and this is true for all other serine residues of chymotrypsin; only ser195 becomes acidic due to the unique constellation of the three amino acid residues because the protein has folded uniquely in space. You may be curious about the importance about an acidic serine residue. The negatively charged oxygen anion is able to make a nucleophilic attack on the carbonyl carbon of the peptide bond of its substrate, loosening it so that a water molecule can hydrolyse the bond (Fig. 5). The specific site of chymotrypsin (recall that the enzyme is specific to aromatic residues) is a large space created within the enzyme active site and lined by hydrophobic residues which therefore only allow bulky aromatic, hydrophobic amino acids to bind. This binding brings the susceptible peptide bond close to the attacking ser195 residue. In chymotrypsinogen, the substrate binding site is blocked and hence the enzyme is inactive. In-situ activation of trypsin involves a proteolytic cut in chymotry p s i n o g e n which results in a conformational change, exposing the substrate binding pocket.
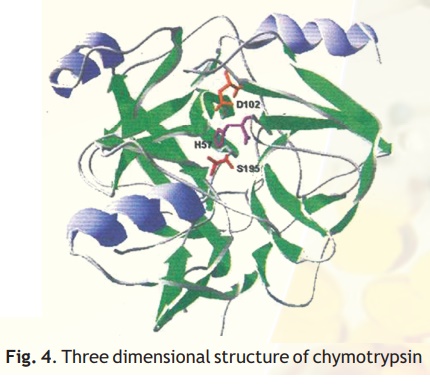
Fig. 4. Three dimensional structure of chymotrypsin
The interesting thing is that when nature has found a useful folding pattern which can cause hydrolysis of protein substrates, it repeats this in a variety of other enzymes. Trypsin, subtilisin (a proteolytic enzyme found in B. subtilis, a bacterium), thrombin (a proteolytic blood clotting factor) and the brain enzyme, acetyl choline esterase all have a reactive serine residue which is central to the catalytic mechanism.
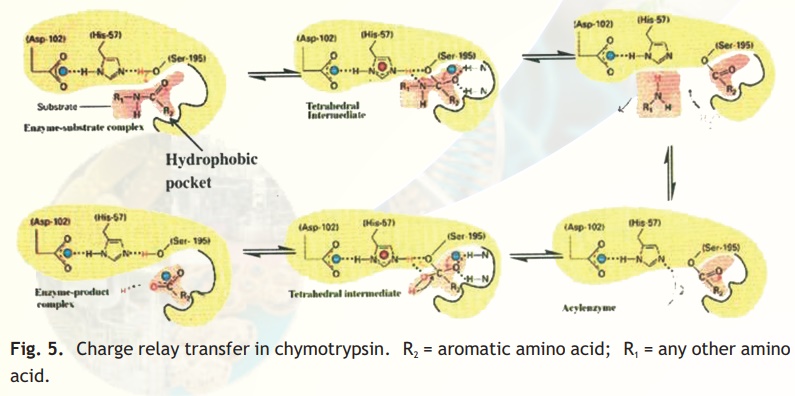
Fig. 5. Charge relay transfer in chymotrypsin. R2 = aromatic amino acid; R1 = any other amino acid.
Certain organophosphate compounds can selectively react with an acidic serine residue thereby knocking off enzyme activity. Nerve gas which was unfortunately used in the first world war had volatile serine alkylating compounds which inactivates the brain enzyme acetyl choline esterase leading to death. Nowadays derivatives of organophosphates such as malathion and parathion which are not toxic to humans are used as mosquito repellants (Mortein, Good Knight) by effecting nerve transmission in insects.
Molecular Disease- Sickle cell anaemia
Sickle cell anaemia is a disease prevalent in parts of Africa and India where malaria is also endemic. The red cells of the patient have a pronounced morphological change and resemble the shape of a farmer’s sickle and thus the name of the disease. Because these unusually shaped red cells have impaired oxygen carrying capacity and further get stuck in the small capillaries they lead to the anaemic conditions observed in patients. Interestingly such sickled RBCs resist malarial infection and hence offer some selection unfortunately for malaria to be co-prevalent with sickle cell anaemia. One of the first attempts to study the molecular basis of sickle cell anaemia was to compare the electrophoretic mobility of normal (Hb) and sickle cell haemoglobin (scHb). On finding that Hb moved faster than scHb, Linus Pauling predicted that the latter differed in a charged amino acid. This was confirmed by V. M. Ingram in 1957 who pioneered a useful technique called protein finger printing in the famous Laboratory of Molecular Biology (LMB) at Cambridge, UK. LMB has been the Mecca for protein sequencing, DNA sequencing, X-ray crystallography, deduction of the Double helix structure of DNA, Hybridoma technology and Nematode developmental studies. Established in 1952 under the leadership of Max Perutz (Received Nobel Prize for the structure of Haemoglobin) this institution has produced 9 Nobel Prize winners.
Protein Finger printing- Peptide Mapping
This technique involves the generation and 2-D analysis of peptides from a protein. Each protein has a unique peptide map (2-D analysis) and hence serves as a fingerprint for the protein. The steps involved in generating a peptide map/fingerprint are as follows (Fig. 6).
1. Pure Hb and scHb are taken separately into test tubes.
2. The Hb and scHb are digested with the proteolytic enzyme trypsin which cleaves the protein after basic amino acid residues Arg and Lys.
3. Two separate strips of Whatman filter paper are spotted with Hb and scHb tryptic peptides and the peptides allowed to separate using the technique of paper electrophoresis at pH 2.0. Highly charged peptides will migrate more towards the anode/cathode.
4. The paper strips are dried, attached to larger squares of Whatman paper and chromatographed at right angles to the electrophoretic direction using a solvent system Butanol: Water:Acetic acid. In such a system peptides will separate based on
their partition coefficient between the solvent and paper which is dependant on the relative hydrophobicity of the peptides. More hydrophobic peptides will move with the solvent to longer distances.
5. The chromatograms are dried and stained with a suitable visualisation reagent like Ninhydrin wherein peptide containing regions appear as orange yellow spots.
6. The peptide map for Hb and scHb are compared and it was found that one peptide was differently placed in the scHb map.
7. On examining this peptide and determining its amino acid sequence, Ingram found that it had a valine substitution for glutamic acid in the peptide.
The single substitution of valine for glutamic acid (val/glu are at the 6th position of the haemoglobin beta chain) dramatically changes the structure of scHb making it form fibres within the RBC resulting in the deformation of the cell (sickling). Since the disease was due to a molecular alteration the term molecular disease was applied.
Peptide mapping became a useful technique to compare similar proteins from different sources. Slowly the information became too vast and computers were used to store this data into databases so that homology searches could be made. The protein fingerprinting data has been further augmented with new databases containing 2-D electrophoresis patterns of entire proteins from a given cell type, a technique developed by O'Farrel.
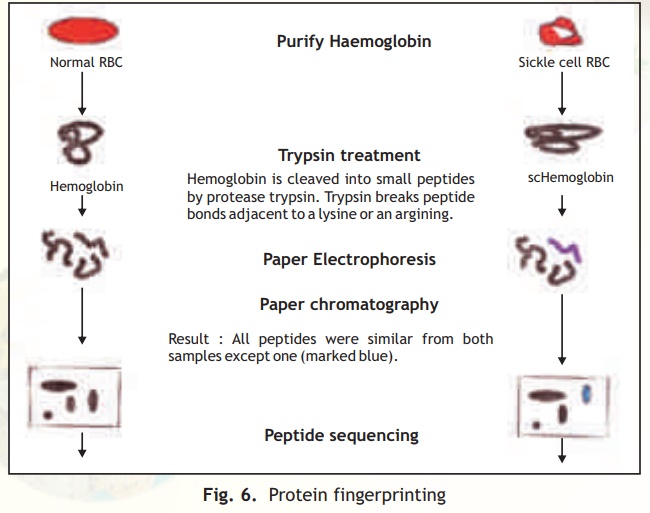
Fig. 6. Protein fingerprinting
2-D Gel Electrophoresis
Two different techniques are combined in this procedure- Isoelectro focussing (IEF) and SDS-PAGE (Fig. 7).
In simple electrophoresis, the mobility of proteins is due to their charge, which is pH dependant. At its isoelectric pH (pI), a protein does not possess any charge and thus will not move in an applied electric field. This feature is exploited in the technique of IEF, which separates proteins on the basis of their different pI values. Usually IEF is performed in thin tube gels. A pH gradient is set up within the IEF gel by the inclusion of polymeric buffers known as ampholytes. These, like proteins have many positive and negative charges and hence varying pIs. Because of their smaller sizes they move rapidly in an electrophoretic run setting up pH gradients when they come to rest at specific distances from the anode/cathode when they have no net charge. A protein sample from a cell or any other source is then electrophoresed within these tubes wherein the different proteins separate and migrate to their pI zones. The tubes containing the separated proteins is then laid on a SDS-PAGE slab gel and electrophoresis continued at right angles to the IEF direction.
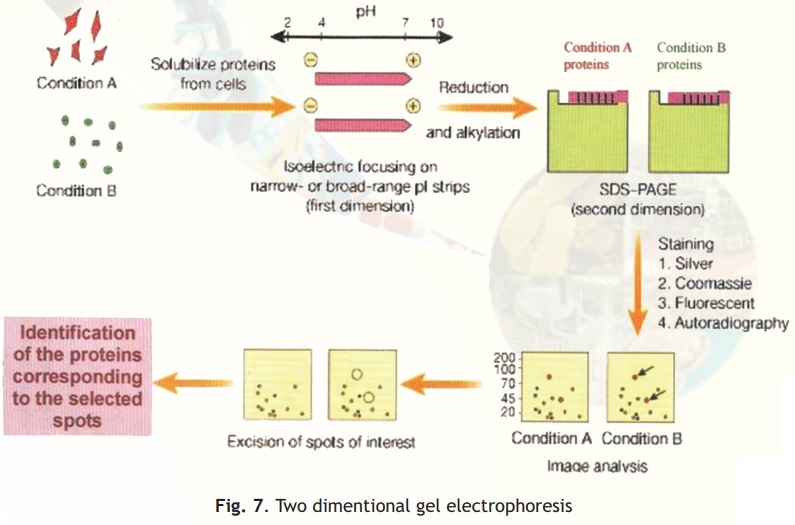
In SDS-PAGE proteins separate on the basis of their size and hence at the end of this electrophoretic run proteins are separated into 2-D patterns with high resolution as two properties of the proteins have been exploited in their separation- charge and size. Proteins in the gels are stained with silver stains or other highly sensitive dyes and can be scanned, and pictures stored into computer databases for analysis.
Related Topics