Chapter: Biotechnology: Protein Structure And Engineering
3-D Shape of Proteins
3-D Shape of Proteins
The morphology, function and activity of a cell are all dependant on the proteins expressed. Proteins perform a variety of roles. The three dimensional properties of proteins have an important bearing on their function. The first step in determining the structure of a protein is to isolate it in a pure form from its cellular location (note bacterial, plant or animal cell). The purified protein is then crystallised so that using a technique called X-ray crystallography its three dimensional structure can be deduced. Nowadays another powerful technique called Nuclear Magnetic Resonance (NMR) has been developed which can deduce protein structures in solution and hence crystallisation is not required. However the protein in either technique has to be purified and some general procedures used to purify proteins will be discussed in subsequent sections. In general when we refer to the structure of a protein this involves two aspects- the chemical structure which is the amino acid sequence of the polypeptide and its folding in space which is referred to its 3-D structure.
One of the major breakthroughs in protein sequence determination was achieved in the middle of the last century by Dr. Frederick Sanger who developed the first sequencing reagent FDNB (fluoro dinitro benzene) and a general strategy for sequencing. By using these methods he was able to sequence the important hormone insulin which is required by diabetics and more importantly he demonstrated for the first time that proteins were linear polymers of amino acids. For this work he was awarded the Nobel Prize and it will be interesting for you to know that several years later he was awarded a second Nobel Prize for developing a sequencing technique for DNA which has been described in the Recombinant DNA technology previously. Another protein chemist, Pehr Edman in 1950 developed another sequencing reagent and procedure which is used in modern day sequenators as the procedure has been automated. Notably using the sequence of insulin established by Sanger a biotechnology company called Eli Lilli was able to develop recombinant human insulin which is the major source for insulin administration to diabetics worldwide.
With the availability of pure proteins, scientists like Linus Pauling, G.N.Ramachandran, Max Perutz and John Kendrew to name a few started developing techniques to study the 3-D shapes of proteins using high resolution X-rays. They laid the foundation for deducing protein structure by enunciating the basic rules which govern protein folding and the forces which cause the folding and stabilise them. Hence from these studies the concepts of planarity of the peptide bond, secondary structures such as alpha helix and beta pleats were developed.
Protein structure has been divided into four hierarchial levels to understand their organisation:
The linear order or sequence of covalently linked amino acid sequence is defined as primary structure. Depending on the nature and arrangement of the amino acids present different parts of the polypeptide chain form secondary structures like alpha helices and beta pleats. The tertiary structure organisation of these secondary structural elements occurs when these get compacted with each other to form compact spherical or globular units which are also thermodynamically stable conformations of these molecules in aqueous solutions (note cytoplasm is mainly water). In compaction several non-covalent interactions occur between the amino acid side chains. Thequarternary structure is the association of two or more independent proteins/polypeptides via non-covalent forces to give a multimeric protein (Fig. 2). The individual peptide units of this protein are referred to as subunits and they may be identical or different from one another.
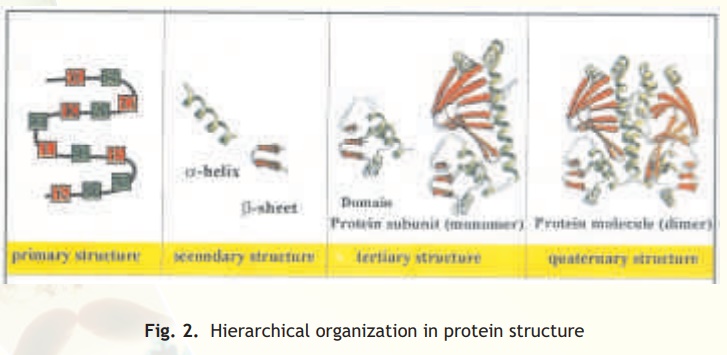
Fig. 2. Hierarchical organization in protein structure
The dominant forces which cause linear protein chains to undergo folding in space lies to a large extent in the chemistry of the amino acid residues they contain. Amino acids are broadly divided into three main groups- polar or hydrophilic (eg. serine, glutamine), charged (eg. aspartate, arginine) and hydrophobic (eg. tryptophan, valine). Hence based on these features amino acid side chains can interact in space by a variety of non-covalent forces which is the basis of forming and stabilising protein structures in space. Let us examine some of the major non-covalent forces found in proteins.
Non-covalent bonds
The non-covalent interactions involved in organising the structure of protein molecules can be broadly divided into four categories:
• Ionic bonds
• Hydrogen bonds
• Van der Waals forces
• Hydrophobic interactions
Ionic bonds
These involve interactions between the oppositely charged groups of a molecule. For example the positively charged amino acid side chains of lysine and arginine can form salt bridges with the negatively charged side chains of aspartate and glutamate. These ionic interactions are also known as salt bridges because these are dominant bonds found in salts like sodium chloride wherein the positively charged sodium ion interacts with the negatively charged chloride ion. However, although ionic bonds have similar strengths to covalent bonds in vacuo, the bond strength of ionic bonds is vastly reduced in water due to the insulating qualities (dielectric strength) of water. Ionic bonds are highly sensitive to pH and salt concentration.
Hydrogen bonds
Hydrogen bonds are formed by "sharing" of a hydrogen atom between two electronegative atoms such as Nitrogen and Oxygen. In this case strongly polarised bonds between hydrogen and a small, very electronegative atom (N,O or F) allow a strong dipole-dipole bond to be formed with another small very electronegative element (N, O or F, Fig. 3). Importantly, the very small sizes of these elements also allow them to approach each other so closely that a partial covalent bond is also formed (e.g.O-H---N). It is to be noted that the partial covalent character means that these bonds (H-bonds) are directional and strongest when the nuclei of all three involved atoms are in a linear arrangement.
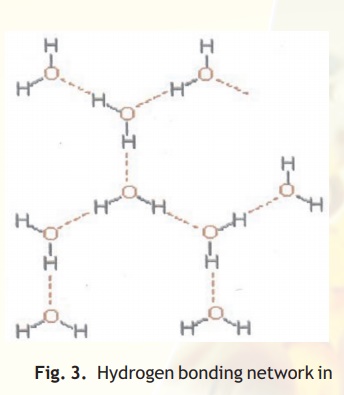
Fig. 3. Hydrogen bonding network in water
Van der Waals forces
These forces are weak attractions (or repulsions) which occur between atoms at close range. The Van der Waals types of forces are essentially contact forces, proportional to the surface areas in contact. These forces are of little significance at a distance due to the rapid 1/r6 (r is the inter-atomic distance) fall off. Even though weak, these bonds can be important in macromolecules because the large surface areas involved can result in reasonably large total forces.
Hydrophobic interactions
Hydrophobic interactions can be best explained by taking an example of oil in water. The oil tends to separate out fairly quickly, not because the oil molecules "want to get together", but because the water forces them out. The hydrophobic interaction is thus a manifestation of hydrogen bonding network in water. In water, each molecule is potentially bonded to four other molecules through H-bonds (Fig. 3).
If a non-polar molecule, which cannot participate in hydrogen bonding, or in electrostatic interactions with water molecules, is added into water , a number of hydrogen bonds will be broken and not replaced. Since hydrogen bonds are favourable interactions, there will be an energy cost to putting non-polar molecules into water. Water therefore forces these molecules out of solution to minimise the surface of contact and thus the number of hydrogen bonds which are broken. Such forces known as hydrophobic forces are among the most important in driving proteins to fold into compact structures (globular) in water. Also, these forces are responsible to make different proteins assemble together to form structures found in muscles, membranes and other organs. In proteins therefore, hydrophobic regions are preferentially located away from the surface of the molecule and form the interior core of the protein.
Related Topics