Chapter: Biotechnology: Protein Structure And Engineering
Characterisation of Proteins
Characterisation of Proteins
Techniques listed below characterise proteins with respect to properties such as mass, isoelectric charge, amino-acid sequence etc. Alongside they can also detect impurities as these are very sensitive techniques requiring small amounts, often micrograms of the sample. The first four
techniques have already been discussed.
1. Electrophoretic techniques, SDS/PAGE.
2. Fingerprinting.
3. Two dimensional gel electrophoresis.
4. Protein sequencing.
5. Mass spectrometry.
Mass spectrometry
Mass spectrometry (MS) has emerged as an important tool in biotechnology. It is extremely useful in obtaining protein structural information such as peptide mass or amino acid sequences. It is also useful in identifying the type and location of amino acid modification within proteins. One of the major attractions of mass spectrometry is that as little as picomoles (10-12) of a protein sample can be analysed. A mass spectrometer is an analytical device that determines the molecular weight of chemical compounds by separating molecular ions according to their mass/charge ratio (m/z) ratios. The molecular ions are generated either by a loss or gain of a charge (e.g. electron ejection, protonation or deprotonation). After the ions are formed they can be separated according to their m/z ratio and finally detected. The process of ionisation, ion separation and detection in a mass spectrum can provide molecular weight or even structural information. A sample M with a molecular weight greater than 1200 D can give rise to multiple charged ions such as (M+nH)n+. Proteins/peptides have many suitable sites for protonation as all the backbone amide nitrogen atoms could be protonated theoretically as well as certain amino acid side chains such as lysine and arginine which contain primary amine functional groups.
A schematic diagram of the various parts of a mass spectrometer is indicated in Fig.10. Basically a vapourised sample of a protein or peptide is introduced into the instrument wherein it undergoes ionisation. The charged molecules are then electrostatically propelled into a mass analyser (filter) which separates the ions according to their m/z ratio. The signal received upon detection of the ions at the detector is transferred to a computer which stores and processes the information.
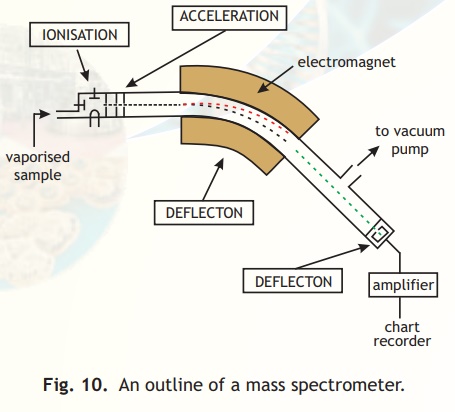
Fig. 10. An outline of a mass spectrometer.
The goal of mass spectrometric analysis of biomolecules like peptides and proteins is to create gas phase ions from polar charged molecules which are generally non-volatile. A popular method called Matrix Assisted Laser Desorption Ionisation (MALDI) is used to volatalise and protonate peptides and proteins. In this procedure, the sample is transferred from a condensed phase to a gas phase with the help of a solid matrix. Ion formation in MALDI is achieved by directing a pulsed laser beam onto a sample suspended or dissolved in a matrix. The matrix plays a key role in this technique by absorbing the laser light energy and causing the matrix material to vaporise. In the gas phase, the matrix plays a role in sample ionisation. The charged molecules are directed by electrostatic lenses from the ionisation source to the mass analyzer.
Multiply charged ions on analysis show patterns as indicated in Fig. from which molecular mass can be deduced as also indicated in the legend below Fig. 11.
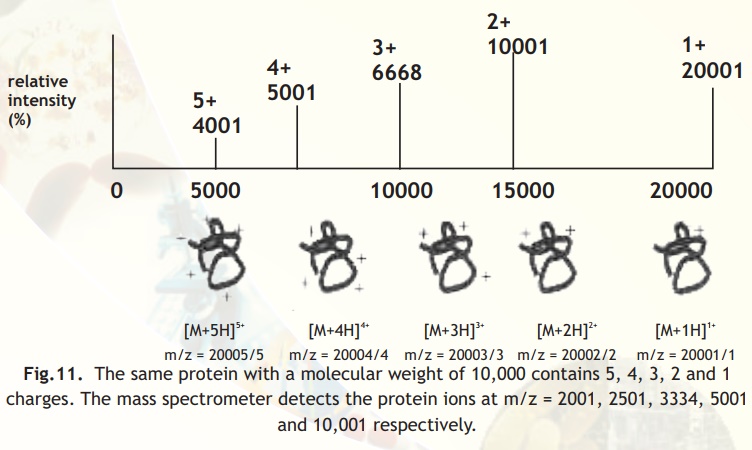
Fig.11. The same protein with a molecular weight of 10,000 contains 5, 4, 3, 2 and 1 charges. The mass spectrometer detects the protein ions at m/z = 2001, 2501, 3334, 5001 and 10,001 respectively.
Typically for protein identification, a crude extract is separated on a 2D gel. After visualising the proteins on the gel, protein spots are excised and used for mass analysis by the MS technique described above. For doing so the protein in the 2D gel after extraction from gel is either used intact or it is cleaved into small peptides with a protease like trypsin which makes the mass analysis easier. These peptides are separated on an on-line liquid chromatography system before introduction into the mass spectrometer. Liquid chromatography techniques like ion exchange, affinity or reverse phase column chromatography can be used to separate the peptides. These peptides are either sequenced directly or the mass of peptides is analysed using database searches. With the human genome sequencing project, it is now possible to identify new proteins by combining the mass spectrometric information with the genomic information using Bioinformatics tools.
Related Topics