Chapter: Basic & Clinical Pharmacology : Adrenoceptor Antagonist Drugs
Specific Agents
SPECIFIC AGENTS
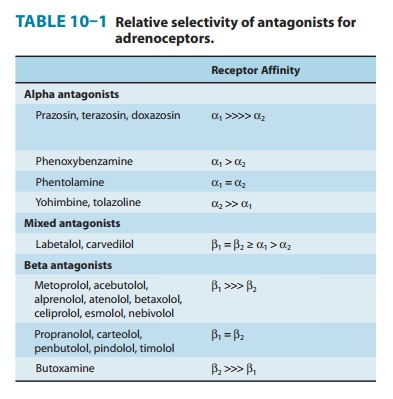
Phenoxybenzamine binds covalently toαreceptors, causing irre-versible blockade of
long duration (14–48 hours or longer). It is somewhat selective for α1 receptors but less so
than prazosin (Table 10–1). The drug also inhibits reuptake of released
norepinephrine by presynaptic adrenergic nerve terminals. Phenoxybenzamine
blocks histamine (H1), acetylcholine, and serotonin receptors as
well as α
receptors .
The
pharmacologic actions of phenoxybenzamine are primarily related to antagonism
of α-receptor–mediated
events. The most significant effect is attenuation of catecholamine-induced
vasocon-striction. While phenoxybenzamine causes relatively little fall in
blood pressure in normal supine individuals, it reduces blood pres-sure when
sympathetic tone is high, eg, as a result of upright pos-ture or because of
reduced blood volume. Cardiac output may be increased because of reflex effects
and because of some blockade of presynaptic α2 receptors in cardiac sympathetic nerves.
Phenoxybenzamine
is absorbed after oral administration, although bioavailability is low and its
kinetic properties are not well known. The drug is usually given orally,
starting with dosages of 10 mg/d and progressively increasing the dose until
the desired effect is achieved. A dosage of less than 100 mg/d is usually
suffi-cient to achieve adequate α-receptor blockade. The major use of
phenoxybenzamine is in the treatment of pheochromocytoma .
Most
adverse effects of phenoxybenzamine derive from its α-receptor–blocking action; the most important
are orthostatichypotension and tachycardia. Nasal stuffiness and inhibition of
ejaculation also occur. Since phenoxybenzamine enters the central nervous
system, it may cause less specific effects, including fatigue, sedation, and
nausea. Because phenoxybenzamine is an alkylating agent, it may have other adverse
effects that have not yet been characterized.
Phentolamine is a potent competitive antagonist at bothα1and α2 receptors (Table
10–1). Phentolamine reduces peripheral resistance through blockade of α1 receptors and
possibly α2 recep-tors on vascular smooth muscle. Its cardiac stimulation
is due to antagonism of presynaptic α2 receptors (leading to enhanced release of
norepinephrine from sympathetic nerves) and sympa-thetic activation from
baroreflex mechanisms. Phentolamine also has minor inhibitory effects at
serotonin receptors and agonist effects at muscarinic and H1 and H2
histamine receptors. Phentolamine’s principal adverse effects are related to
cardiac stimulation, which may cause severe tachycardia, arrhythmias, and
myocardial ischemia. Phentolamine has been used in the treat-ment of
pheochromocytoma. In addition it is sometimes used to reverse local anesthesia
in soft tissue sites; local anesthetics are often given with vasoconstrictors
that slow their removal. Local phentolamine permits reversal at the end of the
procedure. Unfortunately oral and intravenous formulations of phentolamine are
no longer consistently available in the United States.
Prazosin is a piperazinyl quinazoline effective in the
manage-ment of hypertension . It is highly selective for α1 receptors and
typically 1000-fold less potent at α2 receptors. This may partially explain the
relative absence of tachycardia seen with prazosin compared with that of
phentolamine and phenoxyben-zamine. Prazosin relaxes both arterial and venous
vascular smooth muscle, as well as smooth muscle in the prostate, due to
blockade of α1 receptors. Prazosin is extensively metabolized in humans;
because of metabolic degradation by the liver, only about 50% of the drug is
available after oral administration. The half-life is nor-mally about 3 hours.
Terazosin is another reversibleα1-selective antagonist that iseffective in
hypertension ; it is also approved for use in men with urinary symptoms due to
benign prostatic hyperplasia (BPH). Terazosin has high bioavailability but is
extensively metabo-lized in the liver, with only a small fraction of unchanged
drug excreted in the urine. The half-life of terazosin is 9–12 hours.
Doxazosin is efficacious in the treatment of
hypertension andBPH. It differs from prazosin and terazosin in having a longer
half-life of about 22 hours. It has moderate bioavailability and is extensively
metabolized, with very little parent drug excreted in urine or feces. Doxazosin
has active metabolites, although their contribution to the drug’s effects is
probably small.
Tamsulosin is a competitiveα1antagonist with a structurequite different
from that of most other α1-receptor blockers. It has high
bioavailability and a half-life of 9–15 hours. It is metabolized extensively in
the liver. Tamsulosin has higher affinity for α1A and α1Dreceptors than for the α1Bsubtype. Evidence suggests thattamsulosin has
relatively greater potency in inhibiting contraction in prostate smooth muscle versus vascular
smooth muscle compared with other α1-selective antagonists. The drug’s efficacy in
BPH suggests that the α1A subtype may be the most important α sub-type mediating
prostate smooth muscle contraction. Furthermore, compared with other
antagonists, tamsulosin has less effect on standing blood pressure in patients.
Nevertheless, caution is appro-priate in using any α antagonist in patients with diminished
sym-pathetic nervous system function. Patients receiving oral tamsulosin and
undergoing cataract surgery are at increased risk of the intra-operative floppy
iris syndrome (IFIS), characterized by the billow-ing of a flaccid iris,
propensity for iris prolapse, and progressive intraoperative pupillary
constriction. These effects increase the risk of cataract surgery, and
complications are more likely in the ensu-ing 14 days if patients are taking
these agents.
Related Topics