Chapter: Clinical Dermatology: The skin in systemic disease
Some metabolic disorders
Some
metabolic disorders
Amyloidosis
Amyloid
is a protein that can be derived from several sources, including immunoglobulin
light chains and probably keratins. It is deposited in the tissues under a
variety of circumstances and is then usually in com-bination with a P component
derived from the plasma. Systemic amyloidosis of the type that is secondary to
chronic inflammatory disease, such as rheumatoid arthritis or tuberculosis,
tends not to affect the skin. In contrast, skin changes are prominent in
primary systemic amyloidosis, and also in the amyloid asso-ciated with multiple
myeloma. Skin blood vessels infiltrated with amyloid rupture easily, causing
‘pinch purpura’ to occur after minor trauma. The waxy deposits of amyloid,
often most obvious around the eyes, may also be purpuric. Distinct from the
systemic amyloidoses are localized deposits of amyloid. These are uncommon and
usually take the form of macular areas of rippled pigmentation, or of plaques
made up of coalescing papules. Both types are itchy.
Mucinoses
The
dermis becomes infiltrated with mucin in certain disorders.
1 Myxoedema.
In the puffy hands and face of patientswith hypothyroidism.
2 Pretibial myxoedema.
Pink or flesh-coloured mucin-ous plaques are seen on the lower shins, together
with marked exophthalmos, in some patients with hyper-thyroidism. They may also
occur after the thyroid abnormality has been treated.
3
Scleromyxoedema. A diffuse thickening and
papula-tion of the skin may occur in connection with an immu-noglobulin G (IgG)
monoclonal paraproteinaemia.
4 Follicular mucinosis.
In this condition, the infiltratedplaques show a loss of hair. Some cases are
associated with a lymphoma.
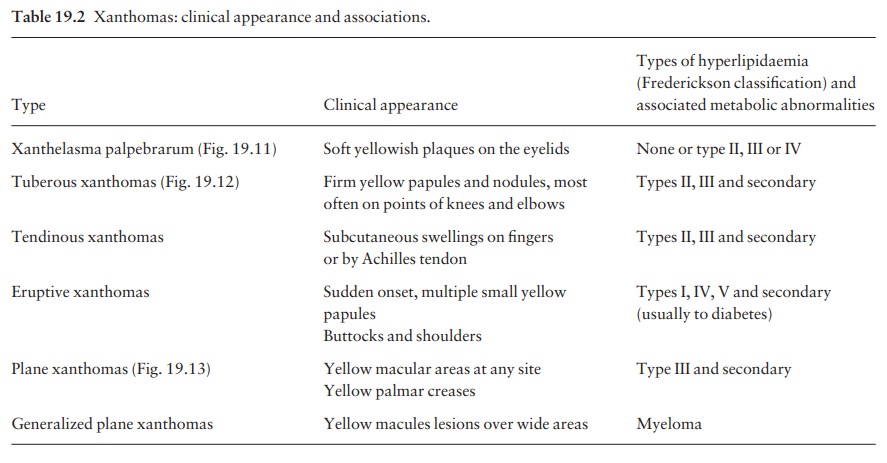
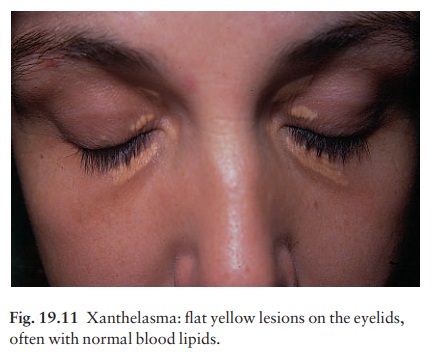
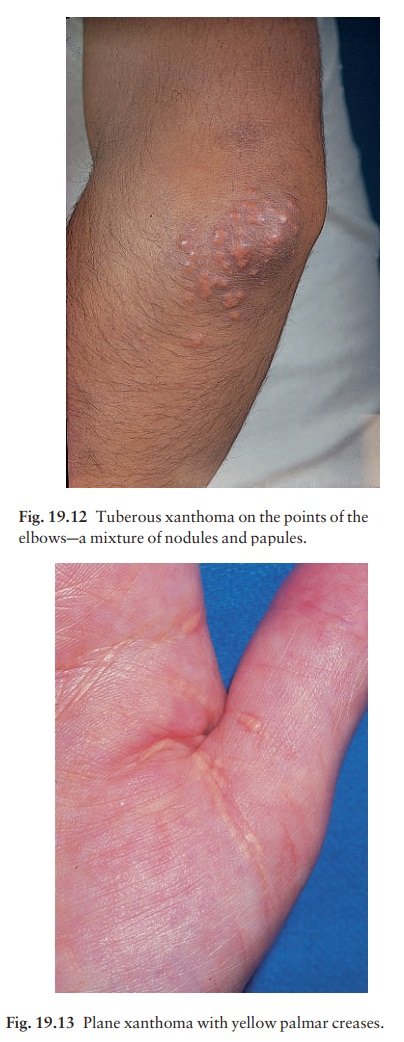
Xanthomas
Deposits
of fatty material in the skin and subcutan-eous tissues (xanthomas) may provide
the first clue to important disorders of lipid metabolism.
Primary
hyperlipidaemias are usually genetic. Theyfall into
six groups, classified on the basis of an ana-lysis of fasting blood lipids and
electrophoresis of plasma lipoproteins. All, save type I, carry an increased
risk of atherosclerosisain this lies their importance and the need for
treatment.
Secondary
hyperlipidaemia may be found in a varietyof diseases including diabetes,
primary biliary cirrhosis, the nephrotic syndrome and hypothyroidism.
The
clinical patterns of xanthoma correlate well with the underlying cause. The
main patterns and their most common associations are shown in Table 19.2.
Phenylketonuria
Phenylketonuria is a rare metabolic cause of hypopig-mentation. Its prevalence is about 1 in 25 000. It is inherited as an autosomal recessive trait, the abnormal gene lying on chromosome region 12q22-q24, and is caused by a deficiency of the liver enzyme phenylalan-ine hydroxylase, which catalyses the hydroxylation of phenylalanine to tyrosine. This leads to the accu-mulation of phenylalanine, phenylpyruvic acid and their metabolites.
Affected individuals have fair skin and hair. They often develop eczema, usually of atopic type, and may be photosensitive.
The accumulation of pheny-lalanine and its metabolites
damages the brain during the phase of rapid development just before and just
after birth. Mental retardation, epilepsy and extrapy-ramidal manifestations
such as athetosis and mental retardation may then occur Oculocutaneous albinism
can usually be distin-guished by its eye signs. The Guthrie test, which detects
raised blood phenylalanine levels, is carried out routinely at birth in most
developed countries.
A
low-phenylalanine diet should be started as soon as possible to prevent further
neurological damage.
Alkaptonuria
In
this rare recessively inherited disorder, based on a homogentisic acid oxidase
deficiency, dark urine may be seen in childhood, and in adult life pigment may
be deposited in various places including the ears and sclera. Arthropathy may
occur.
Fabry’s disease (angiokeratoma corporis diffusum)
A
deficiency of the enzyme α-galactosidase
A is found in this sex-linked disorder (chromosome region Xq21.3–22); abnormal
amounts of glycolipid are deposited in many tissues as a result. The skin
lesions are grouped, almost black, small telangiectatic papules especially
around the umbilicus and pelvis. Progress-ive renal failure occurs in adult
life. Most patients have attacks of excruciating unexplained pain in their
hands. Some female carriers have skin changes, although these are usually less
obvious than those of affected males. Similar skin lesions may be seen in
lysosomal storage disorders such as fucosidosis.
Related Topics