Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Monoclonal Antibodies: From Structure to Therapeutic Application
Pharmacokinetics - Translational Medicine/Development Process
Pharmacokinetics
A thorough and rigorous PK program in the early learning phase of preclinical drug development can provide a linkage between drug discovery and preclinical development. PK information can be linked to PD by mathematical modeling, which allows characterizing the time course of the effect intensity resulting from a certain dosing regimen. Antibodies often exhibit PK properties that are much more complex than those typically associated with small-molecule drugs (Meibohm and Derendorf, 2002). In the following sections we have summarized the basic characteristics of antibody PK.
The PK of antibodies is very different from small molecules. Table 3 summarizes the PK differences between small molecule drugs and therapeutic anti-bodies regarding PK. Precise sensitive and accurate bioanalytical methods are essential for PK interpreta-tion. However, for mAbs, the immunoassays and bioassay methodologies are often less specific as compared to assays used for small molecule drugs (e.g., LC/MS/MS). Monoclonal antibodies are handled by the body very differently than small molecules. In
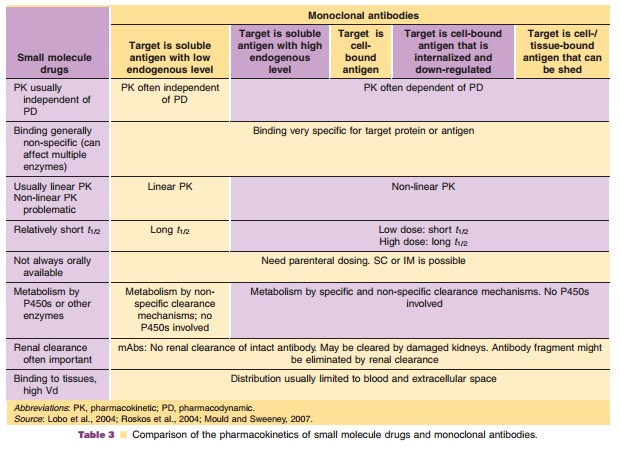
contrast to small molecule drugs, the typical metabolic enzymes and transporter proteins such as cytochrome P450, multi-drug resistance efflux pumps are not involved in the disposition of mAbs. Consequently, drug-drug interaction at the level of these drug metabolizing enzymes and transporters are not com-plicating factors in the drug development process of mAbs and do not need to be addressed by in vitro and in vivo studies. Intact mAbs cannot be cleared by normal kidneys because of the large molecular weight; however, renal clearance processes can play an im-portant role in the elimination of molecules of smaller molecular weight such as Fabs and chemically derived small molecule drugs. The different Absorption,Distribution, Metabolism and Elimination (ADME)processes comprising PK of mAbs will be discussed separately to address their individual specificities.
Absorption
Monoclonal antibodies are not administrated orally because of their limited gastrointestinal stability, low lipophilicity, and size all of which result in insufficient resistance against the hostile proteolytic gastrointest-inal milieu and very limited permeation through the lipophilic intestinal wall. Therefore, intravenous administration is still the most frequently used route, which allows for immediate systemic delivery of large volume of drug product and provides complete systemic availability. Of note, 6 of over 20 FDA approved antibody therapies listed in Table 1 are administered by extravascular route [adalimumab (SC), efalizumab (SC), omalizumab (SC), alefacept (IM), palivizumab (IM), ranibizumab [intravitreal injection (ITV)]. The absorption mechanisms of SC or IM administration are poorly understood. However, it is believed that the absorption of mAbs after IM or SC is likely via lymphatic drainage due to their large molecular weight, leading to a slow absorption rate. The bioavailability for antibodies after SC or IM administration has been reported to be around 50% to 100% with maximal plasma concentrations observed 1 to 8 days following administration (Lobo et al., 2004). For example, following an IM injection, the bioavailability of alefacept was ~60% in healthy male volunteers; its Cmaxwas 3-fold (0.96 vs. 3.1µg/mL) lower and its Tmaxwas 30 times longer (86 vs. 2.8 hours) than a 30-minute IV infusion (Vaishnaw and TenHoor, 2002).
Interestingly differences in PK have also been ob-served between different sites of IM dosing. PAmAb, a fully human mAb against Bacillus anthracis protec-tive antigen, has a significantly different PK between IM-GM (gluteus maximus site) and IM-VL (vastus lateralis site) injection in healthy volunteers (Subramanian et al., 2005). The bioavailability of PAmAb is 50–54% for IM-GM injection and 71% to85% for IM-VL injection (Subramanian et al., 2005). Of note, mAbs appear to have greater bioavailability after SC administration in monkeys than in humans. The mean bioavailability of adalimumab is around 64% with Cmax of 4.7 ± 1.6 µg/mL and Tmax of 131 ± 56 hours after a single 40 mg SC administration in healthy adult subjects (Humira, 2007). The mean bioavailability of omalizumab is around 62% and peak serum concentration was observed at 7 to 8 days after a single SC dose in patients with asthma (Xolair, 2006). Humanized monoclonal anti-interleukin-5 was reported to have completed absorption (118% ± 1.16%) in monkeys following SC administrations (Zia-Amirhosseini et al., 1999). A similar finding was also observed for adalimumab after single SC administra-tion in monkeys (96%).
Distribution
After reaching the bloodstream, mAbs undergo biphasic disposition from serum, beginning with a rapid distribution phase. The distribution volume of the rapid distribution compartment is relatively small in the range of the plasma volume. It is reported that the volume of the central compartment (Vc) is about 2 to 3 L and the steady state volume of distribution (Vss) is around 3.5 to 7 L for mAbs in humans (Lobo et al., 2004; Roskos et al., 2004). The small Vc and Vss for mAbs indicate that the distribution of mAbs is restricted to the blood and extracellular spaces or target tissues, which is in agreement with their hydrophilic nature thus limiting the access to the lipophilic tissue compartments and their large mole-cular weight. Small volumes of distributions are consistent with relatively small tissue: blood ratios for most antibodies typically ranging from 0.1 to 0.5 (Baxter et al., 1994; Berger et al., 2005). For example, the tissue: blood concentration ratios for a murine IgG1 mAb against the human ovarian cancer antigen CA125 in mice at 24 hours after injection is 0.44, 0.39, 0.48, 0.34, 0.10, and 0.13 for the spleen, liver, lung, kidney, stomach and muscle, respectively (Berger et al., 2005). Brain and cerebrospinal fluid are anatomically protected by blood-tissue barriers. Therefore, both compartments are very limited dis-tribution compartments for Abs hindering the access for therapeutic mAbs. For example endogenous IgG levels in CSF were shown to be in the range of only 0.1 to 1% of their respective serum levels (Wurster and Haas, 1994). However, it has been repeatedly pointed out that the reported Vss obtained by traditional non-compartmental or compartmental analysis may be not correct for some mAbs with high extent of catabolism within tissue (Tang et al., 2004; Lobo et al., 2004; Straughn, 2006). The rate and extent of antibody distribution will be dependent on the kinetics of antibody extravasation within tissue, distributionwithin tissue, and elimination from tissue. Convection, diffusion, transcytosis, binding and catabolism are important determining factors for antibody distribu-tion (Lobo et al., 2004). Therefore, the Vss might be substantially greater than the plasma volume in particular for those mAbs demonstrating high binding affinity in the tissue. Effects of the presence of specific receptors (i.e., antigen sink) on the distribution for mAb have been reported by different research groups (Danilov et al., 2001; Kairemo et al., 2001). Danilov et al. (2001) found that anti-PECAM-1 (CD31) mAbs show tissue:blood concentration ratios of 13.1, 10.9, and 5.96 for the lung, liver, and spleen, respectively, in rats at 2 hours after injection. Therefore, the true Vss of the anti-PECAM-1 is likely to be 15-fold greater than plasma volume.
Another complexity, which needs to be consid-ered, is that the distribution via interaction with target proteins (e.g., cell surface proteins) and subsequent internalization of the antigen-mAb complex might be dose-dependent. For the murine analog mAb of efalizumab (M17) (Coffey et al., 2005), a pronounced dose-dependent distribution was demonstrated by comparing tissue:blood concentration ratios for liver, spleen, bone marrow, and lymph node after a tracer dose of radiolabeled M17 and a high dose treatment. The tracer dose of M17 resulted into substantially higher tissue:blood concentration ratios of 6.4, 2.8, 1.6, and 1.3 for the lung, spleen, bone marrow and lymph node, respectively, in mice at 72 hours after injection. Whereas, the saturation of the target antigen at the high dose level reduced the tissue distribution to the target independent distribution and resulted conse-quently into substantially lower tissue:blood concen-tration ratios (less than one).
FcRn may play an important role in the transport of IgGs from plasma to the interstitial fluid of tissue. However, the effects of FcRn on the mAbs’ tissue distribution have not been fully understood. Ferl et al. (2005) reported that a physiologically based pharma-cokinetic (PBPK) model, including the kinetic inter-action between the mAb and the FcRn within intracellular compartments, could describe the bio-distribution of an anti-CEA mAb in a variety of tissue compartments such as plasma, lung, spleen, tumor, skin, muscle, kidney, heart, bone and liver. FcRn was also reported to mediate the IgG across the placental barriers (Junghans, 1997) and the vectorial transport of IgG into the lumen of intestine (Dickinson et al., 1999) and lung (Spiekermann et al., 2002).
Antibody Clearance
Antibodies are mainly cleared by catabolism and broken down into peptide fragments and amino acids which can be recycled-used as energy supply or for new protein synthesis. Due to small molecular weightof antibodies fragments (e.g., Fab and Fv), elimination of these fragments is faster than intact IgGs, and they can be filtered through glomerus and reabsorbed and/or metabolized by proximal tubular cells of the nephron (Lobo et al., 2004). Murine monoclonal anti-digoxin Fab, F(ab0)2 and IgG1 have half-lives of 0.41, 0.70 and 8.10 hr in rats, respectively (Bazin-Redureau et al., 1997). Several studies reported that the kidney is the major route for the catabolism of Fab and elimination of unchanged Fab (Druet et al., 1978; McClurkan et al., 1993).
IgGs have a half-life of approximately 21 days with clearance values of about 3 to 5 mL/day/kg in the clinically used dose range resulting in linear PK. The exception is IgG3, which has only a half-life of 7 days. The half-life of IgGs is much longer than other Igs (IgA 6 days, IgE 2.5 days, IgM 5 days, IgD 3 days). Recent reports have demonstrated that the FcRn receptor is a prime determinant of the disposition of IgG antibodies (Ghetie et al., 1996; Junghans and Anderson, 1996; Junghans, 1997). FcRn, which pro-tects IgG from catabolism and contributes to the long plasma half-life of IgG, was first postulated by Brambell in 1964 (Brambell et al., 1964) and cloned in the late 1980s (Simister and Mostov, 1989a,b). FcRn is a heterodimer comprising of a β2m light chain and a MHC class-I like heavy chain. The receptor is ubiquitously expressed in cells and tissues. Several studies have shown that IgG clearance in β2m knock-out mice (Ghetie et al., 1996; Junghans and Anderson, 1996) and FcRn-heavy chain knockout mice (Roopenian et al., 2003) is increased 10- to 15-fold, with no changes in the elimination of other Igs. Figure 7 illustrates how the FcRn protects IgG from catabolism and contributes to its long half-life. The FcRn binds to IgG in a pH dependent manner: binding to IgG at acidic pH (6.0) at endosome and releasing IgG at physiological pH (7.4). The unbound IgG proceeds to the lysosome and undergoes proteolysis.
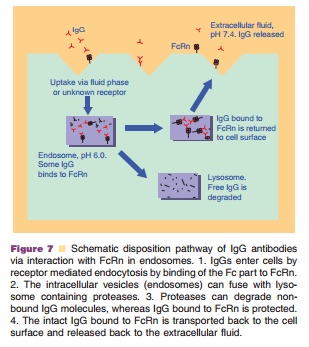
It is demonstrated that IgG half-life is dependent on its affinity to FcRn. The shorter half-life of IgG3 was attributed to its low binding affinity to the FcRn (Junghans, 1997; Medesan et al., 1997). Murine mAbs have serum half-lives of 1 to 2 days in human. The shorter half-life of murine antibodies in human is due to their low binding affinity to the human FcRn receptor. It is reported that human FcRn binds to human, rabbit, and guinea pig IgG, but not to rat, mouse, sheep and bovine IgG; however, mouse FcRn binds to IgG from all of these species (Ober et al., 2001). Interesting, human IgG1 has greater affinity to murine FcRn (Petkova et al., 2006), which indicates potential limitations of using mice as preclinical models for human IgG1 PK evaluations. Ward’s group confirmed that an engineered human IgG1 had disparate properties in murine and human systems (Vaccaro et al., 2006). Engineered IgGs with higher affinity to FcRn have a 2- to 3-fold longer half-life compared with WT in mice and monkeys (Hinton et al., 2006; Petkova et al., 2006). Two engineered human IgG1 mutants with enhanced binding affinity to human FcRn show a considerably extended half-life compared with wild-type in hFcRn transgenic mice (4.35 ± 0.53, 3.85 ± 0.55 days vs. 1.72 ± 0.08 days) (Petkova et al., 2006). Hinton et al. (2006) found that the half-life of IgG1 FcRn mutants with increasing binding affinity to human FcRn at pH 6.0 is about 2.5-fold longer than the WT Ab in monkey (838 ± 187 hours vs. 336 ± 34 hours ).
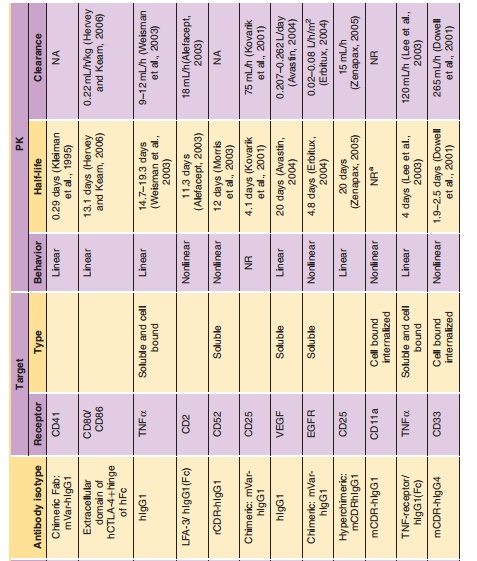
Dose-proportional, linear clearance has been observed for mAbs against soluble antigens with low endogenous levels (such as TNF-α, IFN-α, VEGF and IL-5). For example, linear PK has been observed for a humanized mAb directed to human interleukin-5 following intravenous administration over a 6000-fold dose range (0.05–300 mg/kg) in monkeys (Zia-Amirhosseini et al., 1999). The clearance of rhuMAb against vascular endothelial growth factor after IV dosing (2–50 mg/kg) ranged from 4.81 to 5.59 mL/ day/kg and did not depend on dose (Lin et al., 1999). The mean total serum clearance and the estimated mean terminal half-life of adalimumab was reported to range from 0.012 to 0.017 L/h, 10.0 to 13.6 days, respectively, for a five cohorts clinical trial (0.5–10 mg/ kg), with an overall mean half-life of 12 days (den Broeder et al., 2002). However, mAbs against solubleantigens with high endogenous levels (such as IgE) exhibit non-linear PK. The PK of omalizumab, an antibody against IgE, are linear only at doses greater than 0.5 mg/kg (Petkova et al., 2006; Xolair, 2006).
Elimination of mAbs may also be impacted by interaction with the targeted cell-bound antigen, and this phenomenon was demonstrated by dose-depen-dent clearance and half-life. At low dose, mAbs show a shorter half-life and a faster clearance due to receptor-mediated elimination. With increasing doses, receptors become saturated; the half-life gradually increases to a constant; and the clearance gradually decreases to a constant. The binding affinity (Kd), antigen density and antigen turn-over rate may influence the receptor-mediated elimination. Koon et al. (2006) found a strong inverse correlation between CD25þ cells expression and apparent dacli-zumab (a mAb specifically binding to CD25) half-life. It has been shown that the PK of murine anti-human CD3 antibodies may be determined by the disappear-ance of target antigen (Meijer et al., 2002). In monkeys and mice, clearance of SGN-40, a humanized mono-clonal anti-CD40 antibody, was much faster at low dose, suggesting non-linear PK (Kelley et al., 2006). In addition, Ng et al. (2006) demonstrated that anti-CD4 monoclonal antibody had ~ 5-fold faster CL at 1 mg/ kg dose compared with 10 mg/kg dose (7.8± 0.6 vs. 37.4 ±2.4 mL/day/kg) in healthy volunteers. They also found that receptor-mediated CL contributed to 8.69%, 27.1%, and 41.7% of total CL when the dose was 1, 5, and 10 mg/kg, respectively.
In addition to FcRn and antigen-antibody inter-action, other factors may also contribute to mAb elimination (Lobo et al., 2004; Roskos et al., 2004; Tabrizi et al., 2006):
Immunogenicity of antibody: The elimination of mAbsin humans often increases with increasing level of immunogenicity (Ternant and Paintaud, 2005; Tabrizi et al., 2006).
The degree and the nature of antibody glycosylation: Thestudy conducted by Newkirk et al. (1996) shows that the state of glycosylation of IgG affects the half-life in mice, and that by removing the terminal sugars (sialic acid and galactose), the antibody (IgG2a) will remain in circulation significantly longer. However, a recent study demonstrated that a humanized anti-Aβ mAb with different glycans in the Fc region had the same clearance in mice (Huang et al., 2006).
Susceptibility of antibody to proteolysis: Gillies et al.(2002) improved the circulating half-life of antibody-interleukin 2 immunocytokine 2-fold compared with wild-type (1.0 hour vs. 0.54 hour) by increasing the resistance to intracellular degradation.
Effector function: Effector function, such as interactionswith FcγR, could also regulate elimination and PK of mAbs (Mahmood and Green, 2005). Mutation of the binding site of FcγR has dramatic effects on the clearance of the Ab-IL-2 fusion protein (Gillies et al., 1999).
Concomitant medications: Methotrexate reduced ada-limumab apparent clearance after single dose and multiple dosing by 29% and 44%, respectively in patients with rheumatoid arthritis (Humira, 2007). In addition, azathioprine and mycophenolate mofetil were reported to reduce clearance of basiliximab by approximately 22% and 51%, respectively (Simulect, 2005). These interactions could be explained by the effects of small molecule drugs on the expression of Fcγ receptors. It has been found that methotrexate have the impact on the expression profiles of FcγRI on monocytes significantly in rheumatoid arthritis patients (Bunescu et al., 2004).
Demographics: Body weight, age, disease state, andother factors can also change mAb PK (Mould and Sweeney, 2007) (see discussion on “Population PK”).
Related Topics