Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Monoclonal Antibodies: From Structure to Therapeutic Application
Murine, Chimeric, Humanized and Fully Humanized mAbs - Antibody Structure and Classes
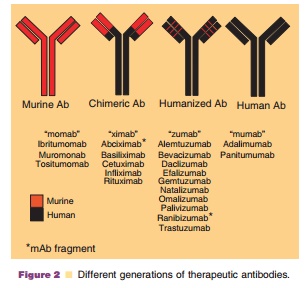
Murine,
Chimeric, Humanized and Fully Humanized mAbs
With the advancement of technology early murine mAbs have been
engineered further to chimeric (mouse CDR human Fc), humanized and fully human
mAbs (Fig. 2). Murine mAbs, chimeric mAbs, huma-nized mAbs and fully humanized
mAbs have 0%, ~60% to ~70%, ~90% to ~95% and ~100% sequencesthat are similar to
human mAbs, respectively. Decreasing the xenogenic portion of the mAb
poten-tially reduces the immunogenic risks of generating anti-therapeutic
antibodies (ATAs). The first thera-peutic mAbs were murine mAbs produced via
hybridomas, however, these murine antibodies easily elicited formation of
neutralizing human anti-mouse antibodies (HAMA) (Kuus-Reichel et al., 1994).
Muromonab-CD3 (Orthoclone OKT3), a first genera-tion mAb of murine origin, has
shown efficacy in the treatment of acute transplant rejection and was the first
mAb licensed for use in humans. It is reported that 50% of the patients who
received OKT3 produced HAMA after the first dose. HAMA interfered with OKT3’s
binding to T-cells, thus decreasing the therapeutic efficacy of the mAb (Norman
et al., 1993). Later, molecular cloning and the expression of the variable
region genes of IgGs have facilitated the generation of engineered antibodies.
A second gen-eration of mAbs, chimeric mAbs consist of human constant regions
and mouse variable regions. The antigen specificity of chimeric mAb is the same
as the parental mouse antibodies; however, the human Fc region renders a longer
in vivo half-life than the parent murine mAb and similar effector functions as
the human Ab. Currently, there are five chimeric antibodies and fragments on
the market (abciximab, basiliximab, cetuximab, infliximab, and rituximab).
These antibodies can still induce human anti-chimeric antibodies (HACA). For
example, about 61% of patients who received infliximab had HACA response
associated with shorter duration of therapeutic effi-cacy and increased risk of
infusion reactions (Baert et al., 2003). The development of ATA is currently
not predictable, as 6 of 17 patients with systemic lupus erythematosus
receiving rituximab developed high-titer HACA (Looney et al., 2004), whereas
only 1 of 166 lymphoma patients developed HACA (McLaughlin et al., 1998).
Humanized mAbs contain significant portions of human sequence except the CDR
which is still of murine origin. There are 10 marketed humanized antibodies on
the market (alemtuzumab, bevacizumab, daclizumab, efalizu-mab, gemtuzumab,
natalizumab, omalizumab, palivi-zumab, ranibizumab and trastuzumab). The
incidence rate of anti-drug antibody [i.e., human anti-human antibody (HAHA)]
was greatly decreased for these humanized mAbs. Trastuzumab has a reported HAHA
incidence rate of only 0.1% (1 of 903 cases) (Herceptin, 2006), but daclizumab had
a HAHA rate as high as 34% (Zenapax, 2005). Another way to achieve
biocompatibility of mAbs is to develop fully humanized antibodies, which can be
produced by two approaches: through phage display library and by using
transgenic XenoMouse with human heavy and light chain gene fragments (Weiner,
2006).
CH2 domain or the hinge region joining CH1 and CH2 have been identified
as the crucial regions for binding to FcγR (Presta
et al., 2002). Engineered mAbs with enhanced or decreased ADCC and CDC activity
have been produced by manipulation of the critical Fc regions. Umana et al.
(1999) engineered an anti-neuroblastomal IgG1 with enhanced ADCC activity
compared with wild-type (WT). Shields et al. (2001) demonstrated that selected
IgG1 variants with im-proving binding to FcγRIIIA
showed an enhancement in ADCC for peripheral blood monocyte cells or natural
killer cells. These findings indicate that Fc-engineered antibodies may have
important applica-tions for improving therapeutic efficacy. It was found that
the FcγRIIIA gene dimorphism generates two allotypes: FcγRIIIa-158V and FcγRIIIa-158F and the polymorphism
in FcγRIIIA is associated with favorable clinical response following rituximab
administration in non-Hodgkin’s lymphoma patients (Cartron et al., 2004;
Dall’Ozzo et al., 2004). Currently, several anti-CD20 mAbs with increased
binding affinity to FcγRIIIA are in clinical trials. The
efficacy of antibody-interleukin-2 fusion protein (Ab-IL-2) was improved by
reducing its interaction with Fcγ
receptors (Gillies et al., 1999). In addition, the Fc portion of mAbs also
binds to the FcRn receptor (FcRn- named based on discovery in neonatal rats as
neonatal Fc receptor), an Fc receptor belonging to the major histocompatibility
complex structure, which is involved in IgG transport and clearance (Junghans,
1997). Engineered mAbs with a decreased or in-creased FcRn binding affinity
have been investigated for the potential of modifying the PK behavior of mAb
(see section “Antibody clearance” for detail).
Related Topics