Chapter: Genetics and Molecular Biology: Advanced Genetic Engineering
Megabase Sequencing
Megabase Sequencing
In addition to mapping, sequencing often provides
fundamental infor-mation for further studies on a gene or gene system. The
sequencing techniques described are adequate for sequenc-ing a few genes, but
one area of great interest, the immune system, possesses hundreds of genes,
many of unknown function. Here hun-dreds of thousands of nucleotides must be
sequenced. Serious effort is now also going into the early steps of determining
the sequence of the entire human genome. Large sequencing projects such as
these require better methods, and several have been developed. The one
described below eliminates the use of radioisotopes and automates the detection
of the bands on gels.
A number of steps in the standard DNA sequencing
procedure seri-ously limit data acquisition. These are obtaining the plasmids
necessary for sequencing the desired region, pouring the gels, exposing and
developing the autoradiograph films, and reading the information from the
films. Several of these steps can be streamlined or eliminated.
Imagine the savings in the Sanger sequencing
technique if each of the four dideoxynucleotides could be tagged with a unique
label. Then, instead of labeling the primer or the first nucleotides
synthesized, the chain terminating nucleotide would possess the label. If this
were done and each of the four labels were distinguishable, the four
dideoxynu-cleotides could be combined in the same synthesis tube and the
complex mixture of the four families of oligonucleotides could be subjected to
electrophoresis in the same lane of the gel. Following electrophoresis, the
four families of oligonucleotides could be distinguished and the entire
sequence read just as though each one occupied a unique lane on the gel.
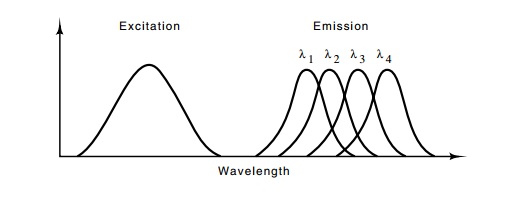
Figure
10.13 Excitation and emission spectra
suitable for DNA sequencing.Each of the four fluorescent groups that emits at
wavelengths λ1, λ2, λ3, and λ4, would be attached to a different base.
Instead of using radioactive label, a fluorescent
label is used. In order that this approach work well, the fluorescent adduct on
the dideoxynu-cleotide must not interfere with the nucleotide’s incorporation
into DNA. Furthermore, each of the four nucleotides must be modified with a
different adduct, one that fluoresces at a different wavelength from the
others. In addition, it is useful if the excitation spectrum of the four
fluorescent molecules substantially overlap so that only one exciting
wavelength is required (Fig. 10.13).
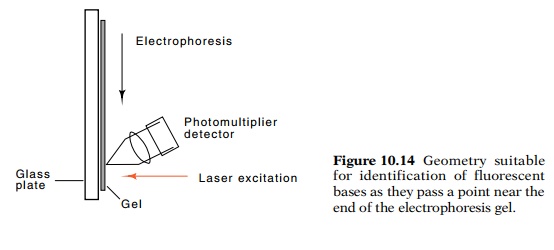
Although the entire gel could be illuminated
following electrophore-sis, it is easier to monitor the passage during
electrophoresis of one band after another past a point near the bottom of the
gel. By measuring the color of the fluorescence passing a point near the bottom
of the gel, the nucleotide terminating this particular size of oligonucleotide
can be determined (Fig. 10.14). One after another, from one nucleotide to the
next, the oligonucleotides pass the illumination point and the color of their
fluorescence is determined, yielding the sequence of the DNA. Multiple lanes
can be monitored simultaneously so that the sequence can be determined
semiautomatically of many different samples simul-taneously. Each lane of such
a gel can provide the sequence of about 400 nucleotides of DNA.
The sensitivity of such a DNA sequencing approach
is less than the radioactive techniques, but it is still sufficiently high that
small DNA samples can be successfully used. A more serious problem than the
sensitivity is the generation of useful samples to be sequenced. One approach
is to generate many random clones from the desired DNA in a vector suitable for
Sanger sequencing, to sequence at least the 300 nucleotides nearest to the
vector DNA, and then to assemble the se-quence of the region by virtue of the
overlaps between various se-quences. This shotgun approach yields the desired
sequences if sufficient clones are available and sufficient time and effort are
ex-pended. In the sequencing of any sizeable amount of DNA, a pure shotgun approach
is not efficient, and great effort is required to close the “statistical” gaps.
When a few gaps remain, it may be easier to close them by chromosome walking
than by sequencing more and more
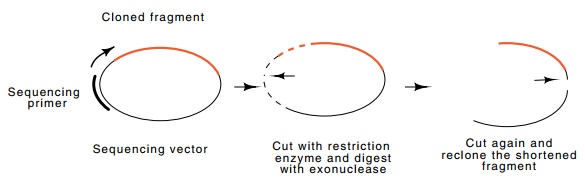
Figure
10.15 Generation of deletions for
sequencing by exonuclease digestion.The vector is opened and digested, then a
fragment is removed by cutting a second time with a restriction enzyme. This
fragment is recloned and sequence is determined by using a primer to a sequence
within the cloning vector adjacent to the location of the inserted fragment. By
performing a series of exonuclease digestions for increasing periods,
progressively larger deletions may be ob-tained.
Another method of generating the necessary clones
for sequencing a large region is to use a nested set of overlapping deletions.
By sequenc-ing from a site within the vector sequences with the use of an
oligonu - cleotide that hybridizes to the vector, the first 400 or so
nucleotides of each of the clones can be determined. The resulting sequences
can easily be assembled to yield the sequence of the entire region.
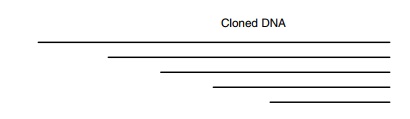
Such a set of clones can be generated by opening a
plasmid containing the cloned DNA, digesting with an exonuclease for various
lengths of time, and recloning so that increasing amounts of the foreign DNA
inserted in the plasmid are deleted (Fig. 10.15).
Related Topics