Chapter: Genetics and Molecular Biology: Advanced Genetic Engineering
Altering Cloned DNA by in vitro Mutagenesis
Altering Cloned DNA by in vitro Mutagenesis
Understanding DNA-related biological mechanisms
requires more than characterizing the DNA and associated proteins. It often
requires altera-tion of the components. Not only does variation of the relevant
parame-ters reveal more about the working mechanism, but the ability to test
variants permits definitive proof of theories. Mutants have been used in
molecular biology almost from its origins, first in the elucidation of
biochemical pathways and now prominently in structural studies of the
mechanisms by which proteins function as enzymes or recognize and bind to
specific nucleotide sequences on DNA.
The efficient isolation of mutations has always
posed a problem in molecular biology. Suppose mutations are desired in a
particular gene or DNA sequence. If the entire organism must be mutagenized,
then to obtain a reasonable number of alterations in the desired target, many
more alterations will inevitably occur elsewhere on the chromosome. Often these
other mutations will be lethal, so the necessary alterations in the target
cannot easily be found. A method is needed for directing mutations just to the
target gene. In vitro mutagenesis of
cloned DNA fragments is a solution to the problem. Only the DNA of the target
sequence is mutagenized. Just this sequence is then put back into cells.
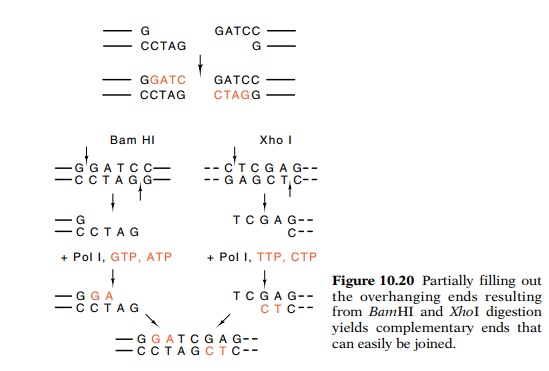
Often random mutations need to be directed to small areas of genes or to specific nucleotides, or specific changes are desired in specific nucleotides. Some changes are easy to make. For
example, insertions and deletions can be generated at the cleavage site of a
restriction enzyme. A four-base insertion can be generated at the cleavage site
of BamHI by filling in the four-base
single-stranded ends with DNA pol Iand ligating the flush ends together.
Similarly, a four-base deletion can be generated by
digesting the single-stranded ends with the single-stranded specific nuclease
S1 be-fore ligation. Variations on these themes are to use DNA pol I in the
presence of only one, two, or three of the nucleotides to fill out part of the
single-stranded ends before nuclease treatment and ligation (Fig. 10.20).
Mixing and matching entire restriction fragments from a region under study is
another closely related method of changing portions of DNA binding sites or
substituting one portion of a protein for another.
More extensive deletions from the ends of DNA
molecules can be generated by double-stranded exonuclease digestion. The
nuclease Bal 31 from the culture medium of the bacterium Alteromonas espejiana is particularly useful for this purpose. With
it, a set of clones with progres-sively larger deletions into a region can
easily be isolated. The addition of linkers after Bal 31 digestion permits
targeted substitution of a set of nucleotides or a change in the number of
nucleotides between two sites. Deletions entering the region from both
directions are isolated. Before recloning, a restriction enzyme linker is
added. After these steps, a pair of deletions can be easily joined via their
linkers to generate a DNA molecule identical to the wild-type except for the
alteration of a stretch comprising the linker (Fig. 10.21). The use of
different pairs of deletions place the linker in different locations so that
the linker can be scanned through a region to determine important areas.
Bases within DNA fragments can be changed with
chemical in vitro mutagenesis.
Hydroxylamine will effectively mutagenize the cytosines in denatured DNA
fragments, which can then be renatured and re-cloned.
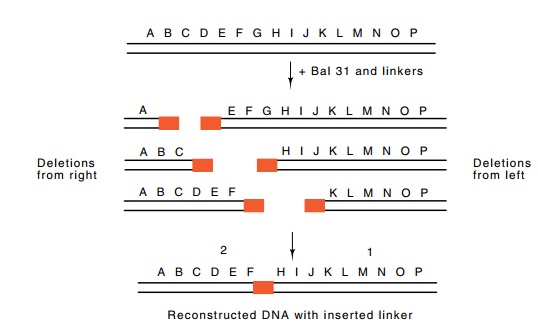
Figure
10.21 Digestion with Bal 31 from either
direction and addition oflinkers generates a set of molecules that can be
rejoined via the linkers to yield a
molecule like the original wild type but with a substitution of some
nucleo-tides.
Alternatively, mutagenesis can be directed to particular regions. One method is to generate a single-stranded region by nicking one strand as a result of digestion with a restriction
enzyme in the presence of ethidium bromide and then briefly digesting with
exonuclease III to generate a gap. The mutagenesis is then performed with a
single-stranded specific reagent such as sodium bisulfite, which mutagenizes
cytosines and ultimately converts them to thymines, or by compelling
misincorporation of bases during repair of a gap.
Figure
10.22 Insertional inactivation of a
yeast gene.
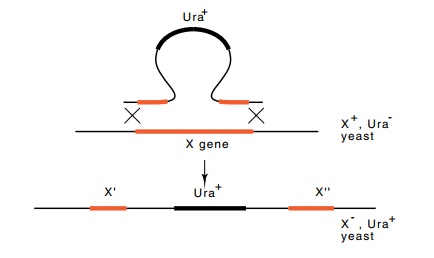
Insertional inactivation can be used to kill
specific genes in yeast (Fig. 10.22). This is a prerequisite to examining the in vivo consequences of mutating the
gene. Suppose that a cloned copy of the gene to be inactivated is available.
Then the central portion of the gene can be replaced by a segment of DNA encoding
one of the genes necessary for the synthesis of uracil. Uracil-requiring yeast
cells are transformed with the segment of DNA containing the gene segments and
the URA region, and selection is performed for cells able to grow without
exogenously added uracil. Since the ends of the transforming DNA segment are
highly recombinogenic, the fragment recombines into the X gene with high
frequency and replaces the former intact copy of the X gene with the damaged
copy. This replacement relieves the uracil requirement of the cells. That the
necessary construct has been generated can be verified by Southern transfers.
Restriction sites flanking the insertion are moved further apart, increasing
the size of this restriction fragment.
When the steps described above are performed on
diploid yeast cells, the result is one chromosome with an insertionally
inactivated copy of the X gene and a second, normal copy of the X gene. To test
whether the X gene is required for growth in haploid cells, haploids containing
the two chromosome types can then be generated by sporulating the diploids. If
the gene with the insertion is completely inviable, only two of the four spores
from each tetrad will be viable.
Related Topics