Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Biophysical and Biochemical Analysis of Recombinant Proteins
Mass Spectrometry - Proteins Analytical Techniques
Mass
Spectrometry
Recent advances in the measurement of the molecular masses of proteins
have made this technique an important analytical tool. While this method was
used in the past to analyze small volatile molecules, the molecular weights of
highly charged proteins with masses of over 100 kilodaltons (kDa) can now be
accurately determined.
Because of the precision of this method, post-translational
modifications such as acetylation or glycosylation can be predicted. The masses
of new protein forms that arise during stability studies provide information on
the nature of this form. For example, an increase in mass of 16 Dalton suggests
that an oxygen atom has been added to the protein as happens when a methionyl
residue is oxidized to a methionyl sulfoxide residue. The molecular mass of
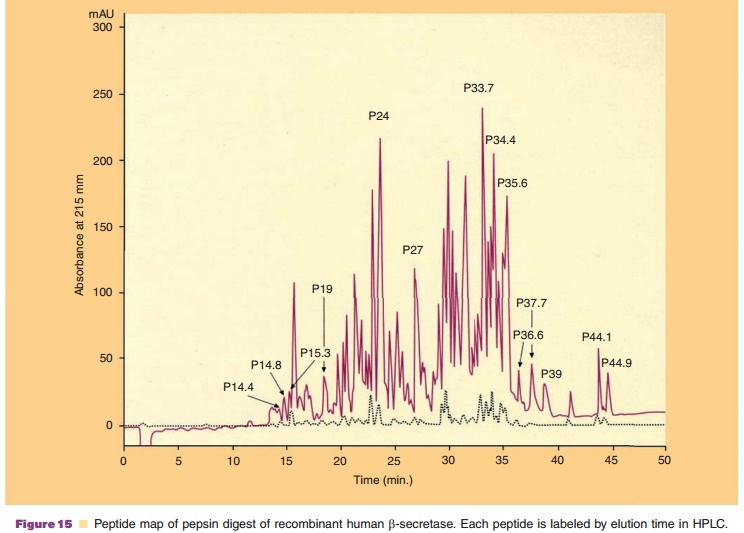
peptides obtained after proteolytic digestion and separation by HPLC indicate
from which region of the primary structure they are derived. Such HPLC
chromatogram is called a “peptide map.” An example is shown in Figure 15. This
is obtained by digesting a protein with pepsin and by subsequently separating
the digested peptides by reverse HPLC. This highly characteristic pattern for a
protein iscalled a “protein fingerprint.” Peaks are identified by elution times
on HPLC. If peptides have molecular masses differing from those expected from
the primary sequence, the nature of the modification to that peptide can be
implicated. Moreover, molecular mass estimates can be made for peptides
obtained from unfractionated pro-teolytic digests. Molecular masses that differ
from expected values indicate that a part of the protein molecule has been
altered, that glycosylation or another modification has been altered, or that
the protein under investigation still contains contaminants.
Another way that mass spectrometry can be used as an analytical tool is
in the sequencing of peptides. A recurring structure, the peptide bond, in
peptides tends to yield fragments of the mature peptide which differ stepwise
by an amino acyl residue. The difference in mass between two fragments
indicates the amino acid removed from one fragment to generate the other.
Except for leucine and isoleucine, each amino acid has a different mass and
hence a sequence can be read from the mass spectrograph. Stepwise removal can
occur from either the amino terminus or carboxy terminus.
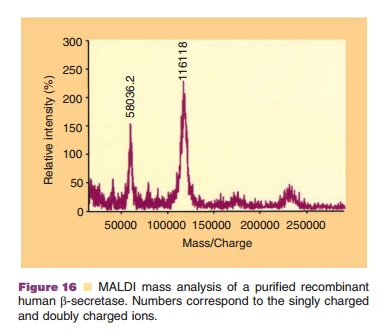
By changing three basic components of the mass spectrometer, the ion source, the analyzer and the detector, different types of measurement may be undertaken. Typical ion sources which volatilize the proteins are electrospray ionization (EI), fast atom bombardment and liquid secondary ion. Common analyzers include quadrupole, magnetic sector, and time of flight instruments. The function of the analyzer is to separate the ionized biomolecules based on their mass to charge ratio. The detector measures a current whenever impinged upon by charged particles. El and matrix-assisted laser desorption (MALDI) are two sources that can generate high molecular weight volatile proteins. In the former method, droplets are generated by spraying or nebulizing the protein solution into the source of the mass spectrometer. As the solvent evaporates, the protein remains behind in the gas phase and passes through the analyzer to the detector. In MALDI, proteins are mixed with a matrix which vaporizes when exposed to laser light, thus carrying the protein into the gas phase. An example of MALDI-mass analysis is shown in Fig. 16, indicating the singly charged ion (116118 Dalton) and the doubly charged ion (58036.2) for a purified protein. Since proteins are multi-charge compounds, a number of components are observed representing mass to charge forms, each differing from the next by one charge. By imputing various charges to the mass to charge values, a molecular mass of the protein can be estimated. The latter step is empirical since only the mass to charge ratio is detected and not the net charge for that particular particle.
Related Topics