Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Biophysical and Biochemical Analysis of Recombinant Proteins
Electrophoresis - Proteins Analytical Techniques
Electrophoresis
Analytical methodologies for measuring protein properties stem from those used in their purification. The major difference is that systems used for analysis have a higher resolving power and detection limit than those used in purification. The two major methods for analysis have their bases in chromato-graphic or electrophoretic techniques.
PAGE
One of the earliest methods for analysis of proteins is PAGE. In this assay, proteins, being amphoteric molecules with both positive and negative charge groups in their primary structure, are separated according to their net electrical charge. A second factor which is responsible for the separation is the mass of the protein. Thus, one can consider more precisely that the charge to mass ratio of proteins determines how they are separated in an electrical field. The charge of the protein can be controlled by the pH of the solution in which the protein is separated. The farther away the protein is from its pI value, that is, the pH at which it has a net charge of zero, the greater is the net charge and hence the greater is its charge to mass ratio. Therefore, the direction and speed of migration of the protein depend on the pH of the gel. If the pH of the gel is above its pI value, then the protein is negatively charged and hence migrates toward the anode. The higher the pH ofthe gel the faster the migration. This type of electro-phoresis is called native gel electrophoresis.
The major component of polyacrylamide gels is water. However, they provide a flexible support so that after a protein has been subjected to an electrical field for an appropriate period of time, it provides a matrix to hold the proteins in place until they can be detected with suitable reagents. By adjusting the amount of acrylamide that is used in these gels, one can control the migration of material within the gel. The more acrylamide, the more hindrance for the protein to migrate in an electrical field.
The addition of a detergent, sodium dodecyl sulfate (SDS), to the electrophoretic separation system allows for the separation to take place primarily as a function of the size of the protein. Dodecyl sulfate ions form complexes with proteins, resulting in an unfold-ing of the proteins, and the amount of detergent that is complexed is proportional to the mass of the protein. The larger the protein, the more detergent that is complexed. Dodecyl sulfate is a negatively charged ion. When proteins are in a solution of SDS, the net effect is that the own charge of the protein is overwhelmed by that of the dodecyl sulfate com-plexed with it, so that the proteins take on a net negative charge proportional to their mass.
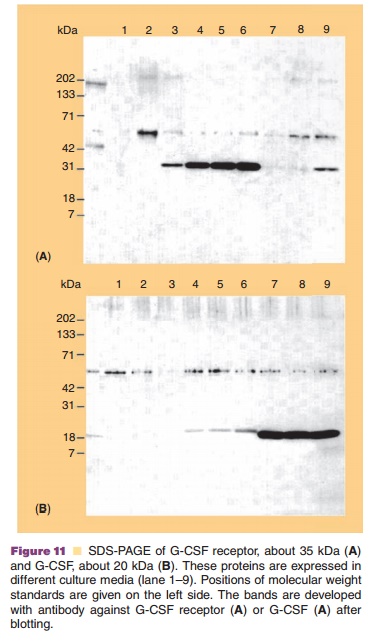
PAGE in the presence of SDS is commonly known as SDS-PAGE. All the proteins take on a net negative charge, with larger proteins binding more SDS but with the charge to mass ratio being fairly constant among the proteins. An example of SDS-PAGE is shown in Figure 11. Here, SDS-PAGE is used to monitor expression of G-CSF receptor and of G-CSF (panel B) in different culture media.
Since all proteins have essentially the same charge to mass ratio, how can separation occur? This is done by controlling the concentration of acrylamide in the path of proteins migrating in an electrical field. The greater the acrylamide concentration, the more difficult it is for large protein molecules to migrate relative to smaller protein molecules. This is some-times thought of as a sieving effect, since the greater the acrylamide concentration, the smaller the pore size within the polyacrylamide gel. Indeed, if the acrylamide concentration is sufficiently high, some high molecular weight proteins may not migrate at all within the gel. Since in SDS-PAGE the proteins are denatured, their hydrodynamic size, and hence the degree of retardation by the sieving effects, is directly related to their mass. Proteins containing disulfide bonds will have a much more compact structure and higher mobility for their mass unless the disulfides are reduced prior to electrophoresis.
As described above, native gel electrophoresis and SDS-PAGE are quite different in terms of the mechanism of protein separation. In native gel electrophoresis, the proteins are in the native state and migrate on their own charges. Thus, this electrophor-esis can be used to characterize proteins in the native state. In SDS-PAGE, proteins are unfolded and migrate based on their molecular mass. As an intermediate case, Blue native electrophoresis is developed, in which proteins are bound by a dye, Coomassie blue, used to stain protein bands. This dye is believed to bind to the hydrophobic surface of the proteins and to add negative charges to the proteins. The dye-bound proteins are still in the native state and migrate based on the net charges, which depend on the intrinsic charges of the proteins and the amounts of the negatively charged dye. This is particularly useful for analyzing membrane proteins, which tend to aggregate in the absence of detergents. The dye prevents the proteins from aggregation by binding to their hydrophobic surface.
Isoelectric Focusing (IEF)
Another method to separate proteins based on their electrophoretic properties is to take advantage of their isoelectric point. In a first run a pH gradient is established within the gel using a mixture of small molecular weight ampholytes with varying pI values. The high pH conditions are established at the site of the cathode. Then, the protein is brought on the gel, e.g., at the site where the pH is 7. In the electrical field the protein will migrate until it reaches the pH on the gel where its net charge is zero. If the protein were to migrate away from this pH value it could gain a charge and migrate toward its pI value again, leading to a focusing effect.
Two-Dimensional Gel Electrophoresis
The above methods can be combined into a procedure called 2-D gel electrophoresis. Proteins are first fractionated by IEF based upon their pI values. They are then subjected to SDS-PAGE perpendicular to the first dimension and fractionated based on the mole-cular weights of proteins. SDS-PAGE cannot be performed before IEF, since once SDS binds to and denatures the proteins they no longer migrate based on their pI values.
Detection of Proteins within Polyacrylamide Gels
Although the polyacrylamide gels provide a flexible support for the proteins, with time the proteins will diffuse and spread within the gel. Consequently, the usual practice is to fix the proteins or trap them at the location where they migrated to. This is accomplished by placing the gels in a fixing solution in which the proteins become insoluble.
There are many methods for staining proteins in gels, but the two most common and well-studied methods are either staining with Coomassie blue or by a method using silver. The latter method is used if increased sensitivity is required. The principle of developing the Coomassie blue stain is the hydro-phobic interaction of a dye with the protein. Thus, the gel takes on a color wherever a protein is located. Using standard amounts of proteins, the amount of protein or contaminant may be estimated. Quantification using the silver staining method is less precise. However, due to the increased sensitivity of this method, very low levels of contaminants can be detected. These fixing and staining procedures denature the proteins. Hence, proteins separated under native conditions, as in native or Blue native gel electrophoresis, will be denatured. To maintain the native state, the gels can be stained with copper or other metal ions.
Capillary Electrophoresis
With recent advances in instrumentation and technol-ogy, capillary electrophoresis has gained an increasedpresence in the analysis of recombinant proteins. Rather than having a matrix, as in PAGE through which the proteins migrate, they are free in solution in an electric field within the confines of a capillary tube with a diameter of 25 to 50 micrometers. The capillary tube passes through an ultraviolet light or fluores-cence detector that measures the presence of proteins migrating in the electric field. The movement of one protein relative to another is a function of the molecular mass and the net charge on the protein. The latter can be influenced by pH and analytes in the solution. This technique has only partially gained acceptance for routine analysis, because of difficulties in reproducibility of the capillaries and in validating this system. Nevertheless, it is a powerful analytical tool for the characterization of recombinant proteins during process development and in stability studies.
Related Topics