Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Biophysical and Biochemical Analysis of Recombinant Proteins
Protein Structure: Secondary Structure
Secondary Structure
a-Helix
Immediately evident in the primary structure of a protein is that each amino acid is linked by a peptide bond. The amide, NH, is a hydrogen donor and the carbonyl, C¼O, is a hydrogen acceptor, and they can form a stable hydrogen bond when they are positioned in an appropriate configuration of the polypeptide chain. Such structures of the polypeptide chain are called secondary structure. Two main structures, α-helix and β-sheet, accommodate such stable hydrogen bonds. The main chain forms a right-handed helix, because only the L-form of amino acids are in proteins, and makes one turn per 3.6 residues. The overall length of α-helices can vary widely. Figure 6 shows an example of a short α-helix. In this case, the C¼O group of residue 1 forms a hydrogen bond to the NH group of residue 5 and C¼O group of residue 2 forms a hydrogen bond with the NH group of residue 6. Thus, at the start of an α-helix, four amide groups are always free and at the end of an α-helix four carboxyl groups are also free. As a result,both ends of an α-helix are highly polar.
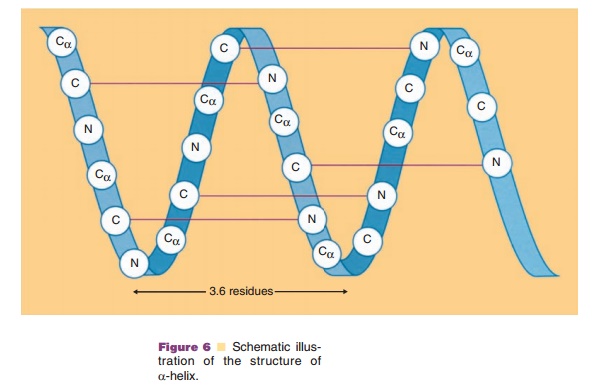
Moreover, all the hydrogen bonds are aligned along the helical axis. Since both peptide NH and C¼O groups have electric dipole moments pointing in the same direction, they will add to a substantial dipole moment throughout the entire α-helix, with the negative partial charge at the C-terminal side and the positive partial charge at the N-terminal side.
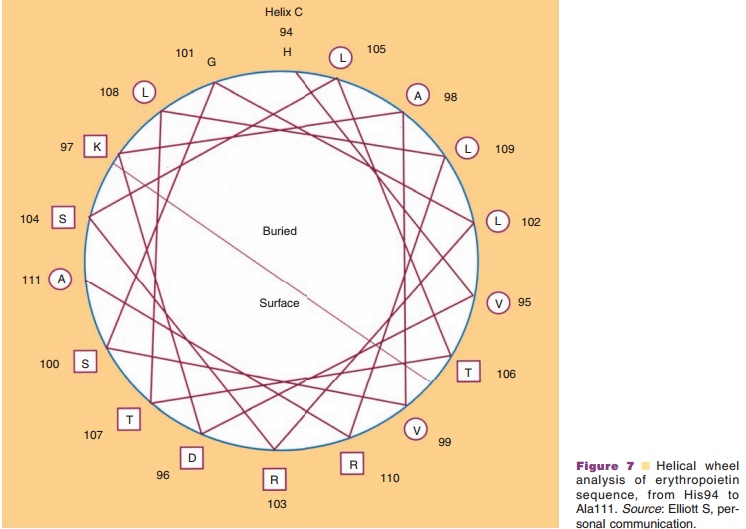
The side chains project outward from the α-helix. This projection means that all the side chainssurround the outer surface of an α-helix and interact both with each other and with side chains of other regions which come in contact with these side chains. These interactions, so-called long-range interactions, can stabilize the α-helical structure and help it to act as a folding unit. Often an α-helix serves as a building block for the three-dimensional structure of globular proteins by bringing hydrophobic side chains to one side of a helix and hydrophilic side chains to the opposite side of the same helix. Distribution of side chains along the α-helical axis can be viewed using the helical wheel. Since one turn in an α-helix is 3.6 residues long, each residue can be plotted every 360L/ 3.6 ¼ 100L around a circle (viewed from the top of α-helix), as shown in Figure 7. Such a plot shows theprojection of the position of the residues onto a plane perpendicular to the helical axis. One of the predicted helices in erythropoietin is shown in Figure 7, using an open circle for hydrophobic side chains and an open rectangle for hydrophilic side chains. It becomes immediately obvious that one side of the α-helix is highly hydrophobic, suggesting that this side forms an internal core, while the other side is relatively hydro-philic and is hence most likely exposed to the surface. Since many biologically important proteins function by interacting with other macromolecules, the information obtained from the helical wheel is extremely useful. For example, mutations of amino acids in the solvent-exposed side may lead to identification of regions responsible for biological activity, while mutations in the internal core may lead to altered protein stability.
β-Sheet
The second major secondary structural element found in proteins is the β-sheet. In contrast to the α-helix, which is built up from a continuous region with a peptide hydrogen bond linking every fourth amino acid, the β-sheet is comprised of peptide hydrogen bonds between different regions of the polypeptide that may be far apart in sequence. β-strands can interact with each other in one of the two ways shown in Figure 8, i.e., either parallel or antiparallel. In a parallel β-sheet, each strand is oriented in the same direction with peptide hydrogen bonds formed be-tween the strands, while in antiparallel β-sheets, the polypeptide sequences are oriented in the opposite direction. In both structures, the C¼O and NH groups project into opposite sides of the polypeptide chain, and hence a β-strand can interact from either side of that particular chain to form peptide hydrogen bonds with adjacent strands. Thus, more than two β-strands can contact each other either in a parallel or in an antiparallel manner,The β-strands which are at the edges of the sheet have unpaired alternating C¼O and NH groups.
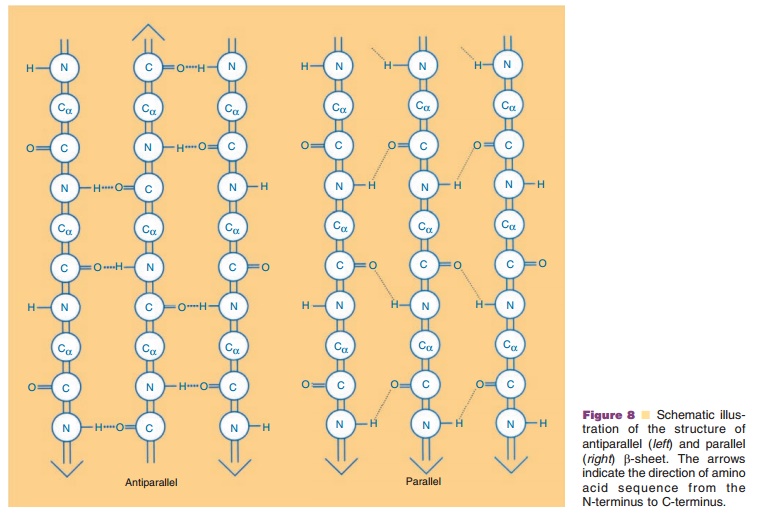
Side chains project perpendicularly to this plane in opposite directions and can interact with other sidechains within the same β-sheet or with other regions of the molecule, or may be exposed to the solvent.
In almost all known protein structures, β-strands are right-handed twisted. This way, the β-strands adapt into widely different conformations. Depending on how they are twisted, all the side chains in the same strand or in different strands do not necessarily project in the same direction.
Loops and Turns
Loops and turns form more or less linear structures, and interact with each other to form a folded three-dimensional structure. They are comprised of an amino acid sequence which is usually hydrophilic and exposed to the solvent. These regions consist of β-turns (reverse turns), short hairpin loops, and longloops. Many hairpin loops are formed to connect two antiparallel β-strands.
As shown in Figure 5A, the amino acid sequences which form β-turns are relatively easy to predict, since turns must be present periodically to fold a linear sequence into a globular structure. Amino acids found most frequently in the β-turn are
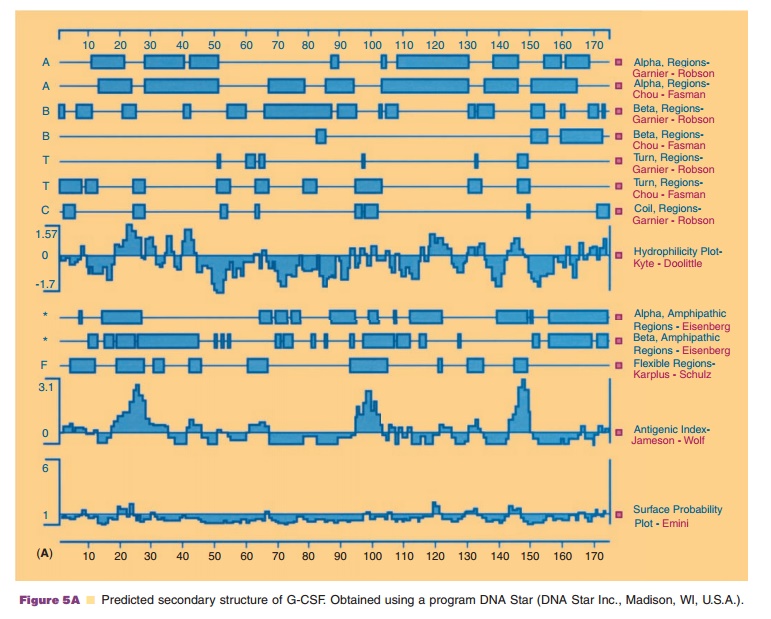
usually not found in α-helical or β-sheet structures. Thus, proline and glycine represent the least observed amino acids in these typical secondary structures. However, proline has an extremely high frequency of occurrence atthe second position in the β-turn while glycine has a high preference at the third and fourth position of a β-turn.
Although loops are not as predictable as β-turns, amino acids with high frequency for β-turns also can form a long loop. Even though difficult to predict, loops are an important secondary structure, since they form a highlysolvent exposed region of the protein molecules and allow the protein to fold onto itself.
Related Topics