Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Biophysical and Biochemical Analysis of Recombinant Proteins
Protein Structure: Primary Structure
PROTEIN
STRUCTURE
Primary Structure
Most proteins which are developed for therapy perform specific functions
by interacting with other small and large molecules, e.g., cell surface
receptors, binding proteins, nucleic acids, carbohydrates and lipids. The
functional properties of proteins are derived from their folding into distinct
three-dimensional structures. Each protein fold is based on its specific
polypeptide sequence in which differ-ent amino acids are connected through
peptide bonds in a specific way. This alignment of the 20 amino acids, called a
primary sequence, has in general all the information necessary for folding into
a distinct tertiary structure comprising different secondary structures such as
α-helices and β-sheets (see below). Because the 20 amino acids possess
different side chains, polypeptides with widely diverse properties are
obtained.
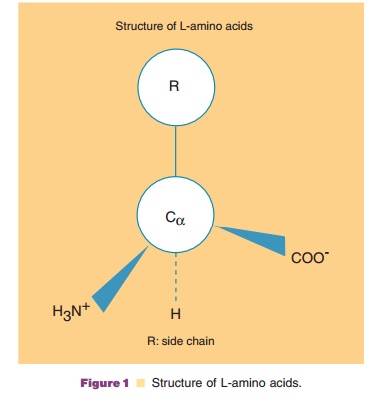
All of the 20 amino acids consist of a Ca carbon to which an amino group, a carboxyl group, ahydrogen, and a side
chain bind in L configuration (Fig. 1). These amino acids are joined by
condensation to yield a peptide bond consisting of a carboxyl group of an amino
acid joined with the amino group of the next amino acid (Fig. 2).
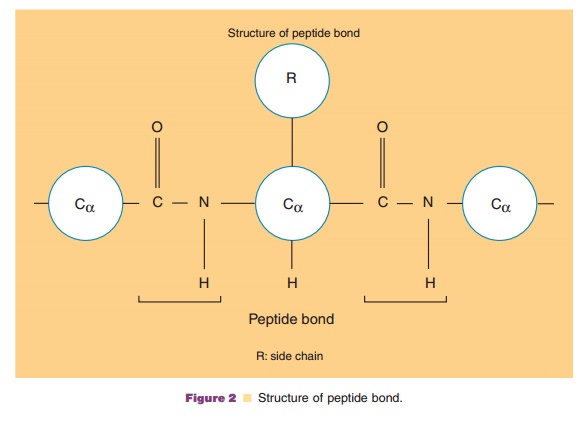
The condensation gives an amide group, NH, at the N-terminal side of Ca, and a carbonyl group, C¼O, at the
C-terminal side. These groups, as well as the amino acyl side chains, play
important roles in protein folding. Due to their ability to form hydrogen
bonds, they make major energetic contributions to the formation of two
important secondary structures, α-helix and β-sheet. The
peptide bonds betweenvarious amino acids are very much equivalent, however, so
that they do not determine which part of a sequence should form an α-helix or
β-sheet. Sequence-dependent secondary structure formation is determined by the
side chains.
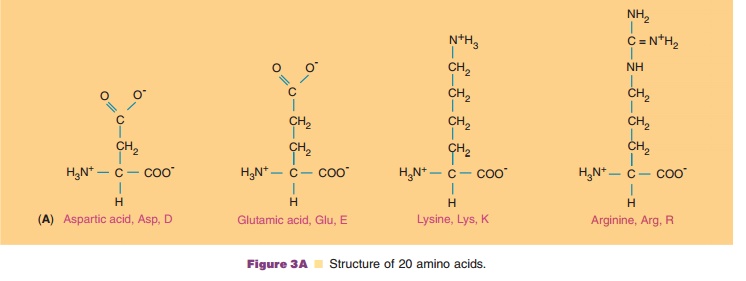
The twenty amino acids commonly found in proteins are shown in Figure 3.
They are described by their full names and three- and one-letter codes. Their
side chains are structurally different in such a way that at neutral pH,
aspartic and glutamic acid are negatively charged and lysine and arginine are
positively charged. Histidine is positively charged to an extent that depends
on the pH. At pH 7.0, on average about half of the histidine side chains are
positively charged. Tyrosine and cysteine are proto-nated and uncharged at
neutral pH, but become negatively charged above pH 10 and 8, respectively.
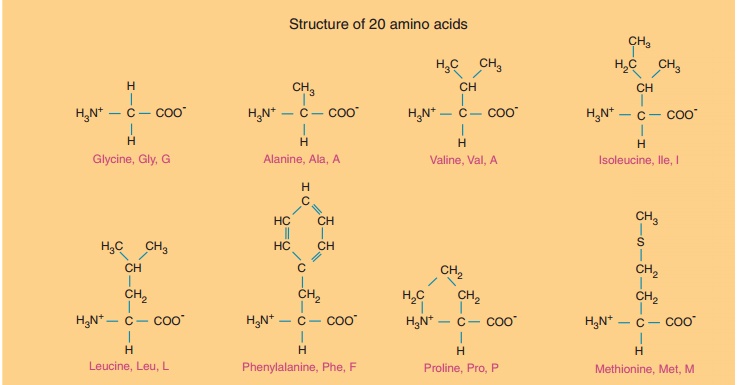
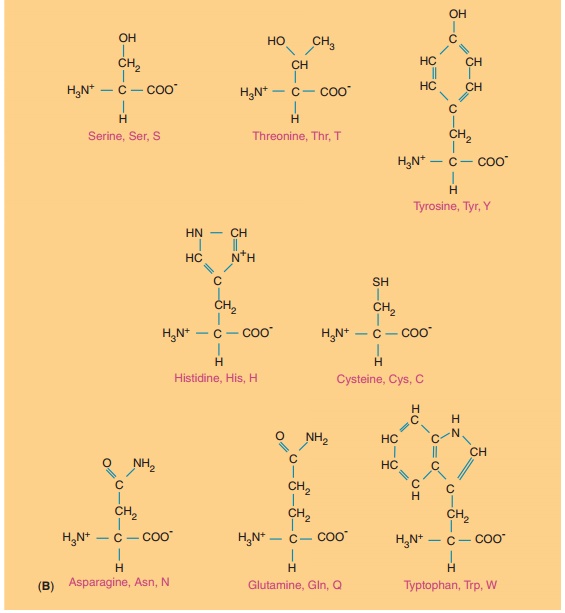
Polar amino acids consist of serine, threonine, asparagine, and
glutamine, as well as cysteine, while nonpolar amino acids consist of alanine,
valine, phenylalanine, proline, methionine, leucine, and iso-leucine. Glycine
behaves neutrally while cystine, the oxidized form of cysteine, is
characterized as hydro-phobic. Although tyrosine and tryptophan often enter
into polar interactions, they are better characterized as nonpolar, or
hydrophobic, as described later.
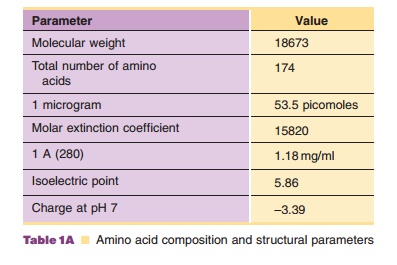
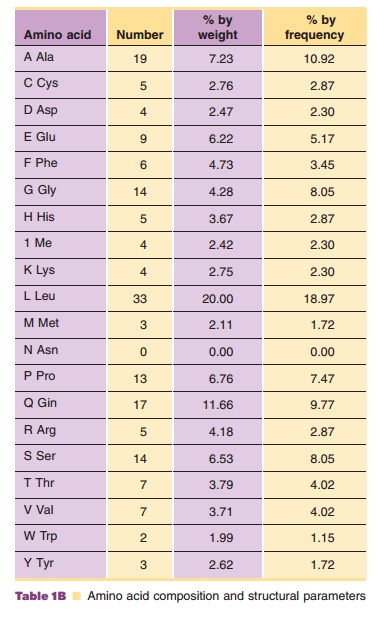
These twenty amino acids are incorporated into a unique sequence based on the genetic code, as the example in Figure 4 shows. This is an amino acid sequence of granulocyte-colony stimulating factor (G-CSF), which selectively regulates proliferation and maturation of neutrophils. Although the exact properties of this protein depend on the location of each amino acid and hence the location of each side chain in the three-dimensional structure, the average properties can be estimated simply from the aminoacid composition, as shown in Table 1; i.e., a list of the total number of each type of amino acid contained in this protein molecule.

Using the pKa values of these side chains and one amino and carboxyl terminus, one
can calculate total charges (positive plus negative charges) and net charges
(positive minus negative charges) of a protein as a function of pH, i.e., a
titration curve. Since cysteine can be oxidized to form a disulfide bond or can
be in a free form, accurate calculation above pH 8 requires knowledge of the
status of cysteinyl residues in the protein. The titration curve thus obtained
is only an approximation, since some charged residues may be buried and the
effective pKa values depend on the local environment of each residue.
Nevertheless, the calcu-lated titration curve gives a first approximation of
the overall charged state of a protein at a given pH and hence its solution
property. Other molecular para-meters, such as isoelectric point (pI; where the
net charge of a protein becomes zero), molecular weight, extinction coefficient,
partial specific volume and hydrophobicity, can also be estimated from the
amino acid composition, as shown in Table 1.
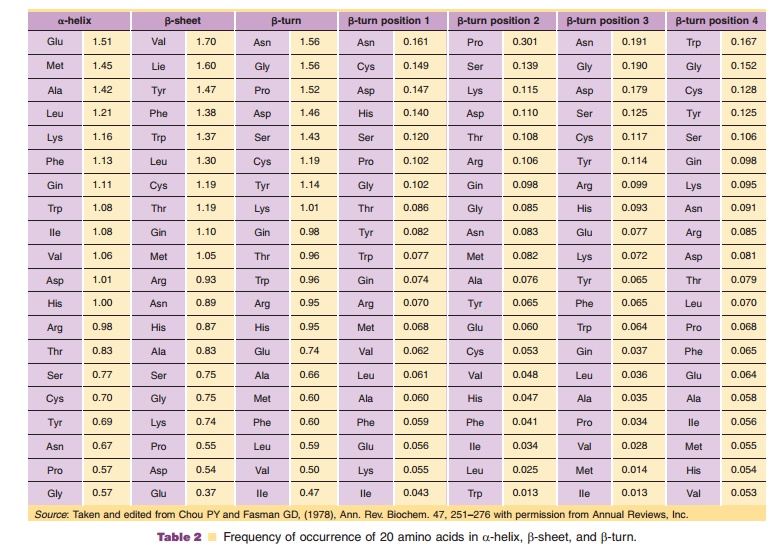
The primary structure of a protein, i.e., the sequence of the twenty amino acids, can lead to the three-dimensional structure because the amino acids have diverse physical properties. First, each type of amino acid has the tendency to be more preferentially incorporated into certain secondary structures. The frequencies with which each amino acid is found in a-helix, β-sheet and β-turn, secondary structures that are discussed later in this chapter, can be calculated as an average over a number of proteins whose three-dimensional structures have been solved. These frequencies are listed in Table 2. The ß-turn has a distinct configuration consisting of four sequential amino acids and there is a strong preference for specific amino acids in these four positions. For example, asparagine has an overall high frequency of occurrence in a ß-turn and is most frequently observed in the first and third position of a ß-turn. This characteristic of asparagine is consistent with its side chain being a potential site of N-linked glycosylation. Effects of glycosylation on the biological and physicochemical properties of proteins are extremely important; however, their contribution to structure is not readily predictable based on the amino acid composition.
Based on these frequencies, one can predict for particular polypeptide
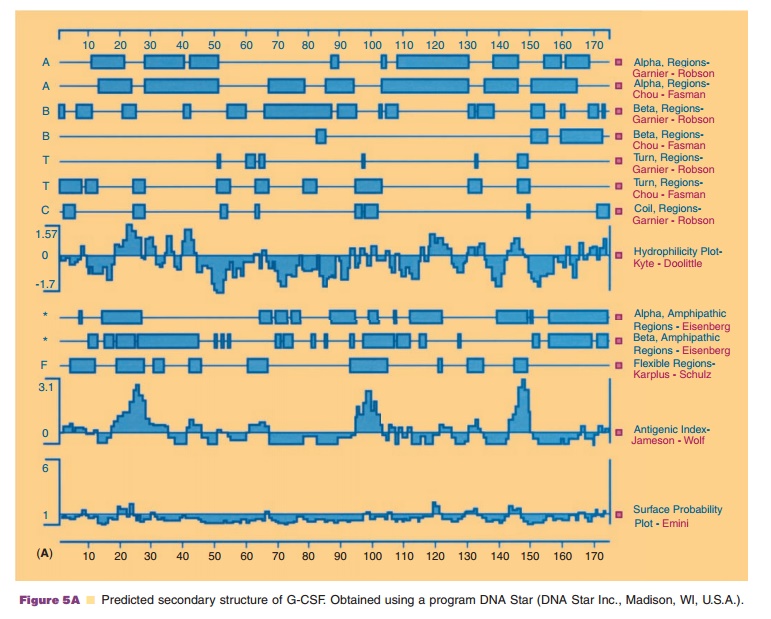
segments which type of secondary structure they are likely to form. As shown in
Figure 5A, there are a number of methods developed to predict the secondary
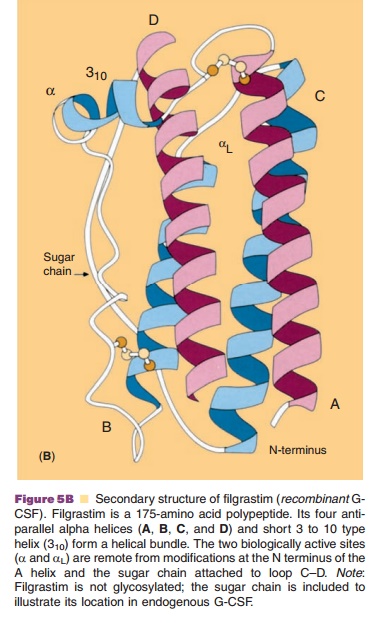
structure from the primary sequence of the proteins. Using G-CSF (Fig. 5B) as
an example, regions of α-helix, ß-sheets, turns, hydrophilicity, and antigen
sites can be suggested.
Another property of amino acids, which impacts on protein folding, is the hydrophobicity of their side chains. Although nonpolar amino acids are basically hydrophobic, it is important to know how hydrophobic they are. This property has been determined by measuring the partition coefficient or solubility of amino acids in water and organic solvents and normalizing such parameters relative to glycine. Relative to the side chain of glycine, a single hydrogen, such normalization shows how strongly the side chains of nonpolar amino acids prefer the organic phase to the aqueous phase. A representation of such measurements is shown in Table 3. The values indicate that the free energy increases as the side chain of tryptophan and tyrosine are transferred from an organic solvent to water and that such transfer is thermodynamically unfavorable. Although it is unclear how comparable the hydrophobic property is between an organic solvent and the interior of protein molecules, the hydrophobic side chains favor clustering together, resulting in a core structure with properties similar to an organic solvent. These hydrophobic character-istics of nonpolar amino acids and hydrophilic characteristics of polar amino acids generate a partition of amino acyl residues into a hydrophobic core and hydrophilic surface, resulting in overall folding.
Related Topics