Chapter: Medical Surgical Nursing: Assessment and Management of Patients With Endocrine Disorders
Management of Patients With Pituitary Disorders
Management of Patients With Pituitary
Disorders
The pituitary gland, or the hypophysis, is a
round structure about 1.27 cm (1Ōüä2 inch) in diameter
located on the inferior aspect of the brain. It is divided into the anterior,
intermediate, and pos-terior lobes.
PITUITARY FUNCTIONAND DYSFUNCTION
Commonly
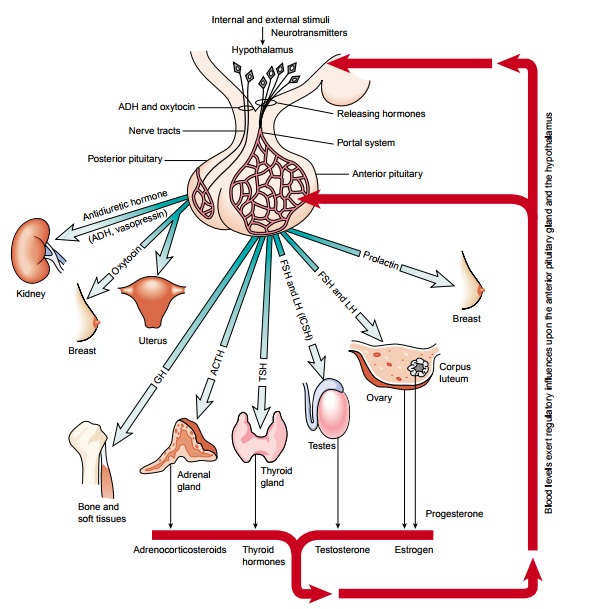
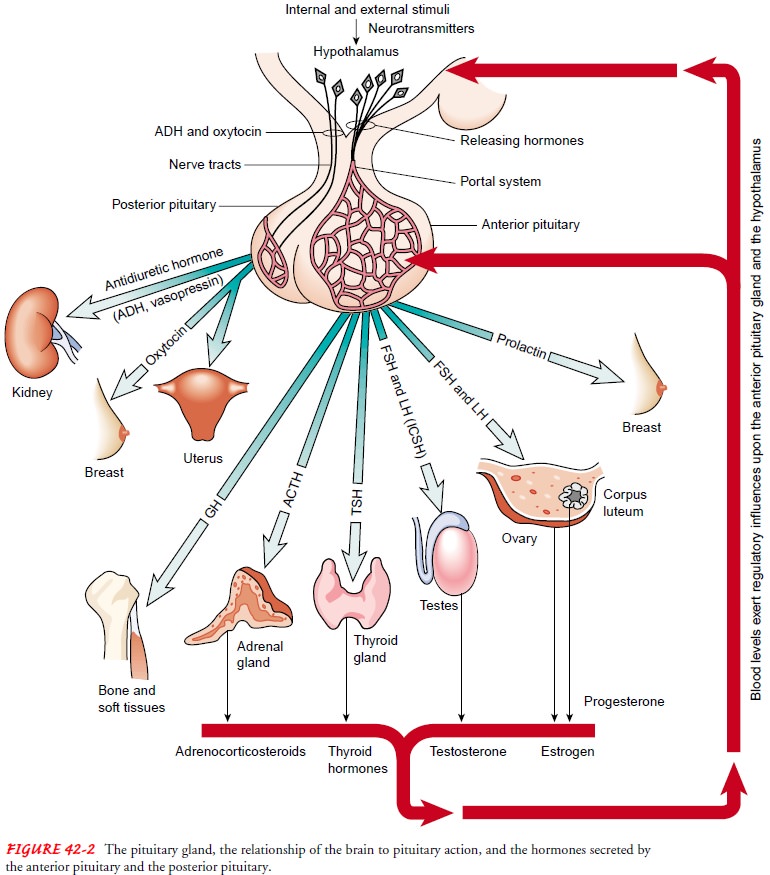
referred to as the master gland, the pituitary secretes hormones that control
the secretion of hormones by other en-docrine glands (Fig. 42-2). The pituitary
itself is controlled by the hypothalamus, an adjacent area of the brain
connected to the pi-tuitary by the pituitary stalk.
Posterior Pituitary
The
important hormones secreted by the posterior lobe of the pi-tuitary gland are vasopressin (antidiuretic hormone
[ADH]) and oxytocin. These hormones
are synthesized in the hypothalamusand travel from the hypothalamus to the
posterior pituitary gland for storage. Vasopressin controls the excretion of
water by the kidney; its secretion is stimulated by an increase in the
osmolal-ity of the blood or by a decrease in blood pressure. Oxytocin
fa-cilitates milk ejection during lactation and increases the force of uterine
contractions during labor and delivery. Oxytocin secre-tion is stimulated
during pregnancy and at childbirth.
Anterior Pituitary
The
major hormones of the anterior pituitary gland are follicle-stimulating hormone
(FSH), luteinizing hormone (LH), pro-lactin, ACTH, thyroid-stimulating hormone
(TSH), and growth hormone (also referred to as somatotropin). The secretion of
these major hormones is controlled by releasing factors secreted by the
hypothalamus. These releasing factors reach the anterior pituitary by way of
the bloodstream in a special circulation called the pituitary portal blood
system. Other hormones include melanocyte-stimulating hormone and
beta-lipotropin; the func-tion of lipotropin is poorly understood.
The
hormones released by the anterior pituitary enter the gen-eral circulation and
are transported to their target organs. The main function of TSH, ACTH, FSH,
and LH is the release of hormones from other endocrine glands. Prolactin acts
on the breast to stimulate milk production. Growth hormone has wide-spread
effects on many target tissues and is discussed later. Hor-mones that stimulate
other organs and tissues are discussed in conjunction with their target organs.
Growth
hormone is a protein hormone that increases protein synthesis in many tissues,
increases the breakdown of fatty acids in adipose tissue, and increases the
glucose level in the blood. These actions of growth hormone are essential for
normal growth, although other hormones, such as thyroid hormone and insulin,
are required as well. Stress, exercise, and low blood glu-cose levels increase
the secretion of growth hormone. The half-life of growth hormone activity in
the blood is 20 to 30 minutes; the hormone is largely inactivated in the liver.
Pathophysiology
Abnormalities
of pituitary function are caused by oversecretion or undersecretion of any of
the hormones produced or released by the gland. Abnormalities of the anterior
and posterior portions of the gland may occur independently. Oversecretion
(hypersecretion) most commonly involves ACTH or growth hormone and results in CushingŌĆÖs syndrome or acromegaly, respectively. Acromegaly,
an excess of growth hormone in adults, results in bone and soft tis-sue
deformities and enlargement of the viscera without an increase in height. In
children, oversecretion of growth hormone results in gigantism, with a person
reaching 7 or even 8 feet tall. Conversely, insufficient secretion of growth
hormone during childhood results in generalized limited growth and dwarfism.
Undersecretion
(hyposecretion) commonly involves all of the anterior pituitary hormones and is
termed panhypopituitarism. In this
condition, the thyroid gland, the adrenal cortex, and the go-nads atrophy
(shrink) because of loss of the trophic-stimulating hormones.
The
most common disorder related to posterior lobe dys-function is diabetes insipidus, a condition in
which abnormally large volumes of dilute urine are excreted as a result of
deficient production of vasopressin.
HYPOPITUITARISM
Hypofunction of the pituitary gland (hypopituitarism) can result from disease of the pituitary gland itself or of the hypothalamus, but the result is essentially the same. Hypopituitarism may result from destruction of the anterior lobe of the pituitary gland. Pan-hypopituitarism (SimmondsŌĆÖ disease) is total absence of all pitu-itary secretions and is rare. Postpartum pituitary necrosis (SheehanŌĆÖs syndrome) is another uncommon cause of failure of the anterior pituitary. It is more likely to occur in women with severe blood loss, hypovolemia, and hypotension at the time of delivery.
Hypopituitarism
is also a complication of radiation therapy to the head and neck area. The
total destruction of the pituitary gland by trauma, tumor, or vascular lesion
removes all stimuli that are normally received by the thyroid, the gonads, and
the adrenal glands. The result is extreme weight loss, emaciation, at-rophy of
all endocrine glands and organs, hair loss, impotence, amenorrhea,
hypometabolism, and hypoglycemia. Coma and death occur if the missing hormones
are not replaced.
PITUITARY TUMORS
Pituitary
tumors are usually benign, although their location and effects on hormone
production by target organs can cause life-threatening effects. Three principal
types of pituitary tumors represent an overgrowth of (1) eosinophilic cells,
(2) basophilic cells, or (3) chromophobic cells (ie, cells with no affinity for
either eosinophilic or basophilic stains).
Clinical Manifestations
Eosinophilic
tumors that develop early in life result in gigantism. The affected person may
be more than 7 feet tall and large in all proportions, yet so weak and
lethargic that he or she can hardly stand. If the disorder begins during adult
life, the excessive skeletal growth occurs only in the feet, the hands, the superciliary
ridge, the molar eminences, the nose, and the chin, giving rise to the clinical
picture called acromegaly. Enlargement, however, involves all tis-sues and
organs of the body. Many of these patients suffer from se-vere headaches and
visual disturbances because the tumors exert pressure on the optic nerves
(Sachse, 2001). Assessment of central vision and visual fields may indicate
loss of color discrimination, diplopia (double vision), or blindness of a
portion of a field of vision. Decalcification of the skeleton, muscular
weakness, and en-docrine disturbances, similar to those occurring in patients
with hyperthyroidism, also are associated with this type of tumor.
Basophilic
tumors give rise to CushingŌĆÖs syndrome with fea-tures largely attributable to
hyperadrenalism, including masculin-ization and amenorrhea in females, truncal
obesity, hypertension, osteoporosis, and polycythemia.
Chromophobic
tumors represent 90% of pituitary tumors. These tumors usually produce no
hormones but destroy the rest of the pituitary gland, causing hypopituitarism.
People with this disease are often obese and somnolent and exhibit fine, scanty
hair, dry, soft skin, a pasty complexion, and small bones. They also experience
headaches, loss of libido, and visual defects pro-gressing to blindness. Other
signs and symptoms include poly-uria, polyphagia, a lowering of the basal metabolic rate, and a subnormal
body temperature.
Assessment and Diagnostic Findings
Diagnostic
evaluation requires a careful history and physical ex-amination, including
assessment of visual acuity and visual fields. Computed tomography (CT) and
magnetic resonance imaging (MRI) are used to diagnose the presence and extent
of pituitary tu-mors. Serum levels of pituitary hormones may be obtained along
with measurements of hormones of target organs (eg, thyroid, adrenal) to assist
in diagnosis if other information is inconclusive.
Medical Management
Surgical
removal of the pituitary tumor through a transsphenoidal approach is the usual
treatment. Stereotactic radiation therapy, which requires use of a
neurosurgery-type stereotactic frame, may be used to deliver external-beam
radiation therapy precisely to the pituitary tumor with minimal effect on
normal tissue. Other treatments include conventional radiation ther-apy,
bromocriptine (dopamine antagonist), and octreotide (syn-thetic analog of
growth hormone). These medications inhibit the production or release of growth
hormone and may bring about marked improvement of symptoms. Octreotide
(Sandostatin) may also be used preoperatively to improve the patientŌĆÖs clinical
condi-tion and to shrink the tumor.
SURGICAL MANAGEMENT: HYPOPHYSECTOMY
Hypophysectomy, or removal
of the pituitary gland, may be per-formed to treat primary pituitary gland
tumors. It is the treat-ment of choice in patients with CushingŌĆÖs syndrome due
to excessive production of ACTH by a tumor of the pituitary gland.
Hypophysectomy may also be performed on occasion as a pallia-tive measure to
relieve bone pain secondary to metastasis of ma-lignant lesions of the breast
and prostate.
Several
approaches are used to remove or destroy the pituitary gland: surgical removal
by transfrontal, subcranial, or oronasalŌĆō transsphenoidal approaches or
irradiation or cryosurgery. Even if surgery succeeds at removing the tumor,
many of the features or symptoms of acromegaly will be unaffected (Sachse,
2001).
The
absence of the pituitary gland alters the function of many body systems.
Menstruation ceases and infertility occurs after total or near-total ablation
of the pituitary gland. Replacement therapy with corticosteroids and thyroid
hormone is necessary; .
DIABETES INSIPIDUS
Diabetes
insipidus is a disorder of the posterior lobe of the pitu-itary gland
characterized by a deficiency of antidiuretic hormone (ADH), or vasopressin.
Great thirst (polydipsia) and large vol-umes of dilute urine characterize the
disorder. It may be sec-ondary to head trauma, brain tumor, or surgical
ablation or irradiation of the pituitary gland. It may also occur with infec-tions
of the central nervous system (meningitis, encephalitis, tu-berculosis) or
tumors (eg, metastatic disease, lymphoma of the breast or lung). Another cause
of diabetes insipidus is failure of the renal tubules to respond to ADH; this
nephrogenic form may be related to hypokalemia, hypercalcemia, and a variety of
med-ications (eg, lithium, demeclocycline [Declomycin]).
Clinical Manifestations
Without
the action of ADH on the distal nephron of the kidney, an enormous daily output
of very dilute, water-like urine with a specific gravity of 1.001 to 1.005
occurs. The urine contains no abnormal substances such as glucose and albumin.
Because of the intense thirst, the patient tends to drink 2 to 20 liters of
fluid daily and craves cold water. In the hereditary form of diabetes
in-sipidus, the primary symptoms may begin at birth. In adults, the onset of
diabetes insipidus may be abrupt or insidious.
The
disease cannot be controlled by limiting fluid intake be-cause the high-volume
loss of urine continues even without fluid replacement. Attempts to restrict
fluids cause the patient to ex-perience an insatiable craving for fluid and to
develop hyper-natremia and severe dehydration.
Assessment and Diagnostic Findings
The
fluid deprivation test is carried out by withholding fluids for 8 to 12 hours
or until 3% to 5% of the body weight is lost. The patient is weighed frequently
during the test. Plasma and urine osmolality studies are performed at the
beginning and end of the test. The inability to increase the specific gravity
and osmolality of the urine is characteristic of diabetes insipidus. The
patient continues to excrete large volumes of urine with low specific grav-ity
and experiences weight loss, rising serum osmolality, and ele-vated serum
sodium levels. The patientŌĆÖs condition needs to be monitored frequently during
the test, and the test is terminated if tachycardia, excessive weight loss, or
hypotension develops.
Other
diagnostic procedures include concurrent measure-ments of plasma levels of ADH
(vasopressin) and plasma and urine osmolality, a trial of desmopressin
(synthetic vasopressin) therapy and intravenous infusion of hypertonic saline
solution. When the diagnosis is confirmed and the cause is not obvious (eg,
head injury), the patient is carefully assessed for tumors that may be causing
the disorder.
Medical Management
The
objectives of therapy are (1) to replace ADH (which is usu-ally a long-term
therapeutic program), (2) to ensure adequate fluid replacement, and (3) to
identify and correct the underlying intracranial pathology. Nephrogenic causes
require different man-agement approaches.
PHARMACOLOGIC THERAPY
Desmopressin
(DDAVP), a synthetic vasopressin without the vascular effects of natural ADH,
is particularly valuable because it has a longer duration of action and fewer
adverse effects than other preparations previously used to treat the disease.
It is ad-ministered intranasally; the patient sprays the solution into the nose
through a flexible calibrated plastic tube. One or two admin-istrations daily
or every 12 to 24 hours usually control the symp-toms (Tierney, McPhee, &
Papadakis, 2001).
Another
form of therapy is the intramuscular administration of ADH, or vasopressin
tannate in oil, which is used when the in-tranasal route is not possible. It is
administered every 24 to 96 hours. The vial of medication should be warmed or
shaken vigorously before administration. The injection is administered in the
evening so that maximum results are obtained during sleep. Abdominal cramps are
a side effect of this medication. Rotation of injection sites is necessary to
prevent lipodystrophy.
Clofibrate,
a hypolipidemic agent, has been found to have an antidiuretic effect on
patients with diabetes insipidus who have some residual hypothalamic vasopressin.
Chlorpropamide (Dia-binese) and thiazide diuretics are also used in mild forms
of the disease because they potentiate the action of vasopressin. The patient
receiving chlorpropamide should be warned of the possi-bility of hypoglycemic
reactions.
If the
diabetes insipidus is renal in origin, the previously de-scribed treatments are
ineffective. Thiazide diuretics, mild salt depletion, and prostaglandin
inhibitors (ibuprofen, indomethacin, and aspirin) are used to treat the
nephrogenic form of diabetes insipidus.
Nursing Management
The
patient with possible diabetes insipidus needs encourage-ment and support while
undergoing studies for a possible cranial lesion. The nurse needs to inform the
patient and family about follow-up care and emergency measures. The nurse also
needs to provide specific verbal and written instructions, show the patient how
to administer the medications, and observe return demon-strations as
appropriate. The nurse also advises the patient to wear a medical
identification bracelet and to carry medication and in-formation about this
disorder at all times. Vasopressin must be administered with caution if the
patient has coronary artery dis-ease because the medication causes
vasoconstriction.
SYNDROME OF INAPPROPRIATE ANTIDIURETIC HORMONE SECRETION
The syndrome of inappropriate antidiuretic
hormone (SIADH)secretion includes excessive growth hormone (ADH)
secretionfrom the pituitary gland even in the face of subnormal serum
os-molality. Patients with this disorder cannot excrete a dilute urine. They
retain fluids and develop a sodium deficiency known as dilu-tional hyponatremia. SIADH is often of nonendocrine origin;
forinstance, the syndrome may occur in patients with bronchogenic carcinoma in
which malignant lung cells synthesize and release ADH. SIADH has also occurred
with severe pneumonia, pneu-mothorax, and other disorders of the lungs, in
addition to malig-nant tumors that affect other organs (Terpstra &
Terpstra, 2000).
Disorders
of the central nervous system, such as head injury, brain surgery or tumor, and
infection, are thought to produce SIADH by direct stimulation of the pituitary
gland. Some medications (vincristine, phenothiazines, tricyclic
antidepressants, thiazide diuretics, and others) and nicotine have been implicated
in SIADH; they either directly stimulate the pituitary gland or increase the
sensitivity of renal tubules to circulating ADH.
Eliminating
the underlying cause, if possible, and restricting fluid intake are typical
interventions for managing this syndrome. Because retained water is excreted
slowly through the kidneys, the extracellular fluid volume contracts and the
serum sodium concentration gradually increases toward normal. Diuretics (eg,
furosemide [Lasix]) may be used along with fluid restriction if severe
hyponatremia is present.
Close
monitoring of fluid intake and output, daily weight, urine and blood
chemistries, and neurologic status is indicated for the patient at risk for
SIADH. Supportive measures and ex-planations of procedures and treatments
assist the patient to deal with this disorder (Terpstra & Terpstra, 2000).
Related Topics