Chapter: Biotechnology Applying the Genetic Revolution: Aging and Apoptosis
Links between Cancer and Aging
LINKS
BETWEEN CANCER AND AGING
The third major factor that triggers
cellular senescence is the presence of oncogenic mutations, that is, mutations
that promote cancer. To defend the organism, tainted cells sacrifice themselves
by starting the genetic program for senescence or apoptosis, thus entering
premature senescence . This brings us to the question of why cells have a deliberate
senescence program. The most prevalent theory is simply to avoid cancer.
There are three key pieces of evidence
for this theory. First, malignant tumors have cells that are immortal and grow
indefinitely because they have bypassed the normal senescence program. Second,
oncogenes that induce a normal cell to become malignant often bypass or block
senescence, thus extending the life span of the cell. Third, loss of p53 or retinoblastoma
protein (pRb) prevents a cell from entering senescence. Many cancers have
mutations in one or both of these two proteins, which are both involved in
control of the cell cycle.
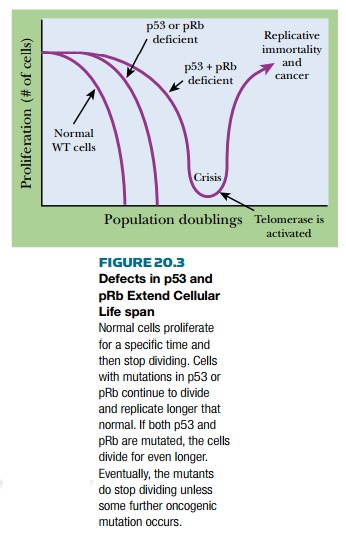
If either of p53 or pRb is defective,
cells replicate for more cycles than usual ( Fig. 20.3 ). If cells are
defective for both p53 and pRb, the effect is additive and the replicative life
span is longer than with either mutation alone. Nonetheless, such double
mutants do eventually enter senescence, after an extended number of cell divisions.
Thus cells have other mechanisms to trigger senescence. The p53 and pRb
proteins are also implicated as key regulators of cellular senescence because
they are the targets for several viral oncogenes. p53 activates several
cellular programs in response to stress, including cellular senescence,
apoptosis, growth arrest, and DNA repair. Its expression is tightly regulated,
because both a surplus and a shortage of p53 are detrimental. The p53 protein
is a transcription factor that regulates its own gene as well as a variety of
other genes. In particular p53 activates transcription of the genes for two other
regulators, PUMA (p53-upregulated modulator of apoptosis) and SLUG. PUMA is a
member of the Bcl-2 protein family that activates Bax, thus promoting apoptosis
(see later section on Mammaliann Apoptosis). Conversely, SLUG protein blocks
the transcription of PUMA and opposes apoptosis. Thus, the balance between PUMA
and SLUG determines the choice between trying to save a damaged cell via growth
arrest plus DNA repair or eliminating the cell via apoptosis. The role of p53
in the death or survival of a damaged cell is very complex and is still being
investigated.
Related Topics