Chapter: Medicine and surgery: Haematology and clinical Immunology
Haemophilia A - Clotting disorders
Clotting disorders
Haemophilia A
Definition
An inherited coagulation disorder resulting from factor VIII deficiency.
Incidence
1–2 in 10,000 two thirds of whom have severe disease.
Age
Inherited, age at presentation depends on severity.
Sex
X linked; males only affected.
Aetiology
Mutations on the X chromosome including deletions, frame shifts and insertions. One third of cases are new mutations.
Pathophysiology
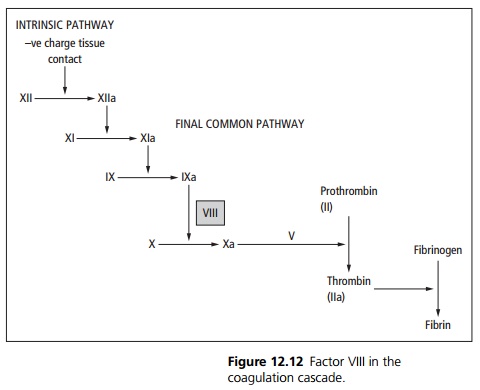
The factor VIII complex is a cofactor in the intrinsic pathway’s activation of factor X (see Fig. 12.12). VIII:c is coded for on the X chromosome and it is this part of the complex that is deficient.
Clinical features
As there are various mutations there is a resultant spectrum of clinical severity dependent on the level of normal VIII complex that is formed:
· Factor VIII levels of less than 1% present within the first 2 years of life with recurrent spontaneous bleeding.
· Factor VIII levels of less than 5% result in severe bleeding following injury although spontaneous haemorrhage does occur.
· Factor VIII levels above 5% causes bleeding only following trauma or surgery, they may not present until later life.
Sites of bleeding include haemarthroses (bleeding into a joint causing pain and restriction of movement), muscle bleeds (often quadriceps, iliopsoas or forearm), intracranial bleeding (presenting as headache, vomiting and lethargy), haematuria and mucosal bleeding.
Complications
There has been significant HIV and Hepatitis C transmitted in factor VIII concentrates during the 1980s, recombinant factor VIII is now the treatment of choice. About 10% of patients develop antibodies to factor VIII:c making them very difficult to treat.
Investigations
Activated partial thromboplastin time is raised, but correctable with 50% normal serum (ie not due to an inhibitor of coagulation) other coagulation measures are normal.
Factor VIII:c assay is low, Factor VIII:vwf is normal.
Management
Bleeding is treated by intravenous administration of factor VIII concentrates. Prophylaxis is the mainstay of treatment for those severely affected to prevent recurrent haemarthroses and permanent joint damage. Patients who develop antibodies to factor VIII concentrates may be successfully treated with recombinant factor VIIa.
1desamino-8-D-arginine vasopressin (DDAVP) releases factor VIII from endothelial cells. The response is proportional to the pretreated factor VIII levels and thus is useful in mild disease.
Carrier detection is possible and antenatal diagnosis uses chorionic villus sampling or fetal blood sampling. Genetic counselling should be offered to patients and carriers.
Related Topics