Chapter: Pharmaceutical Drug Analysis: Gas Liquid Chromatography (GLC)
Gas Liquid Chromatography (GLC): Theory
THEORY
There are, in fact, three
theories that have gained virtually wide recognition and acceptance in
describ-ing a gas chromatographic separation, namely :
(a) Plate
theory,
(b) Rate
theory, and
(c) Random walk
and nonequilibrium theory.
These different theories will be discussed briefly in the
sections that follows :
1. PLATE THEORY
Martin and Synge* first proposed the ‘plate theory’ in 1941, whereby they
merely compared the GC separation to fractional distillation. Thus, the
‘theoretical’ plate is the portion of the column wherein the solute is in
complete equilibrium with the mobile and the stationary phase.
This equilibrium is represented by the following
expression :
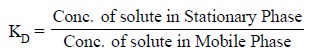
where, KD
= Distribution coefficient.
Thus, the distribution of a solute after ‘n’ equilibrium (plates) may be defined
by the expansion of the binomial in Eq. (a)
below :
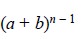 .........................(a)
.........................(a)
where, (n – 1)
= Number of transfers between the plates,
a = 1/(KD + 1), and
b = KD/(KD + 1).
2. RATE THEORY
As the ‘Plate
Theory’ has two serious limitation, viz.,
first : it does not speak of the separating power of a definite length of
column, and second : it does not suggest means of improving the performance of
the column ; the ‘Rate Theory’ has
been introduced which endeavours to include the vital fact that-‘the mobile-phase flows continuously,
besides the solute molecules are constantly being transported and partitioned
in a gas chromatographic column’. It is usually expressed by the following
expression :
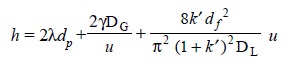 .......................(b)
.......................(b)
where, u = Average linear gas
velocity,
λ= Measure of the packing irregularities
dp = Particle diameter,
γ= Tortuosity factor,
DG = Coefficient of gaseous diffusion of the
solute in the carrier gas,
k′ = Ratio of the amount of
solute in the stationary phase to that in the gas phase, df = Film thickness (usually in µm), and
DL = Diffusion constant of solute in liquid
phase.
Eq. (b) was
first advocated by Van Deemter* in 1956, and may be rewritten as given below
[Eq. (c)] wherein all terms except ‘u’ are constant :
 ........................(c)
........................(c)
3 RANDOM WALK AND NONEQUILIBRIUM THEORY
Giddings** in 1958, first proposed the captioned theory
wherein he suggested a chromatographic separation in terms of a random walk.
Based on a statistical concept the virtual spreading of a ‘solute band’ may be considered by virtue of molecular diffusion, mass transfer, and Eddy diffusion (i.e., flow
pattern effects) were equated to
standard deviation. In fact, this particular approach correlates the spreading
of a chromatographic peak to various
parameters, for instance : mass transfer, diffusion coefficient DG,
particle diameter, velocity of mobile-phase, and finally the length of column.
Thus, coefficient DG, particle diameter, velocity of mobile-phase,
and finally the length of column. Thus, the plate height ‘h’ employing the random walk approach may be expressed as in Eq. (d) :
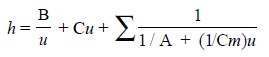 .....................(d)
.....................(d)
where, Cm =
Resistance to mass transfer in gas phase should be treated independently,
A = Eddy diffusion,
B = Longitudinal molecular diffusion in both mobile and
stationary phases, and
C = Kinetic or mass transfer term originating in the
stationary phase.
In actual practice, there are two basic considerations that prevail upon in gas chromatography,
namely :
(a) Retention : The phenomena affecting
retention or hold up on the column, sometimes referred to as the thermodynamic
effect, and
(b) Column Efficiency : The phenomena
affecting column efficiency or the kinetic aspect that governs the tendency for
a particular solute band to ‘broaden’
as it traverses through the column.
However, the resolution or extent of separation of any
two peaks from a column is solely dependent upon both retention and column
efficiency.
Although separations may be caused by elution, frontal
and displacement analyses, yet the elution technique is the most common. This
method makes use of a stream of carrier-gas flowing through the column.
Precisely, a sample is injected into the carrier-gas as a ‘plug’ of vapour that is swept into the head of the packed
chromatographic column. Separation of components that comprise the sample
results from a difference in the multiple forces by which the column materials
tend to retain each of the components.
Irrespective of the nature of the retention that is due
to adsorption, solubility, chemical binding, polarity or molecular filtration,
the column does retain some components longer than others. When in the gas
phase the components are moved toward the column outlet, they are selectively
retarded by the station-ary phase. Consequently, all components pass through
the column at varying speeds and emerge in the inverse order of their retention by the column materials. The
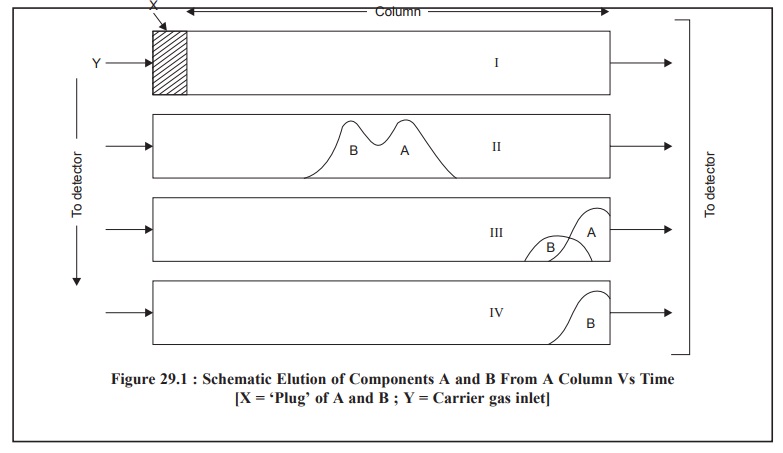
aforesaid process may be outlined schematically
as shown in Figure 29.1
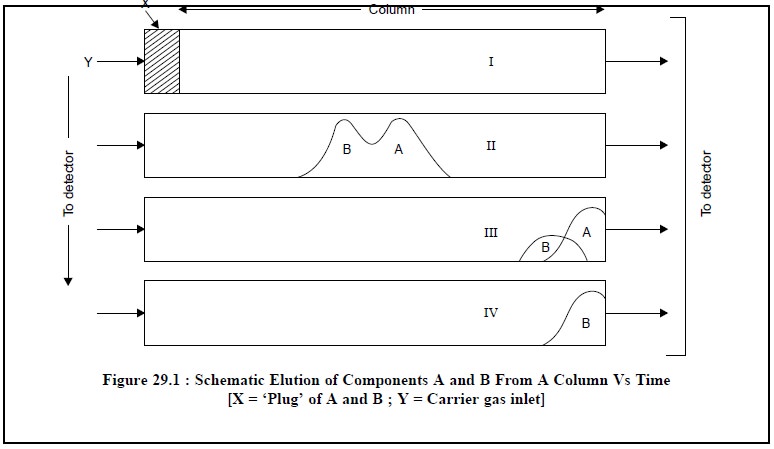
Upon emerging from the column, the gaseous phase
immediately enters a ‘detector’ attached to the column. Here, the individual
components register a series of signals that appear as a succession of peaks
above a base line on the chromatogram.
From Figure 29.1, it is evident that ‘plug’ of mixture A
and B just enters the column 1 at time T1, moves to the middle of
the column I at time T2, part of A has passed through the column III
at time T3 and finally A has passed completely and part of B passed
through the column IV at time T4.
Related Topics