Chapter: Pharmaceutical Drug Analysis: Gas Liquid Chromatography (GLC)
Gas Liquid Chromatography (GLC): Instrumentation
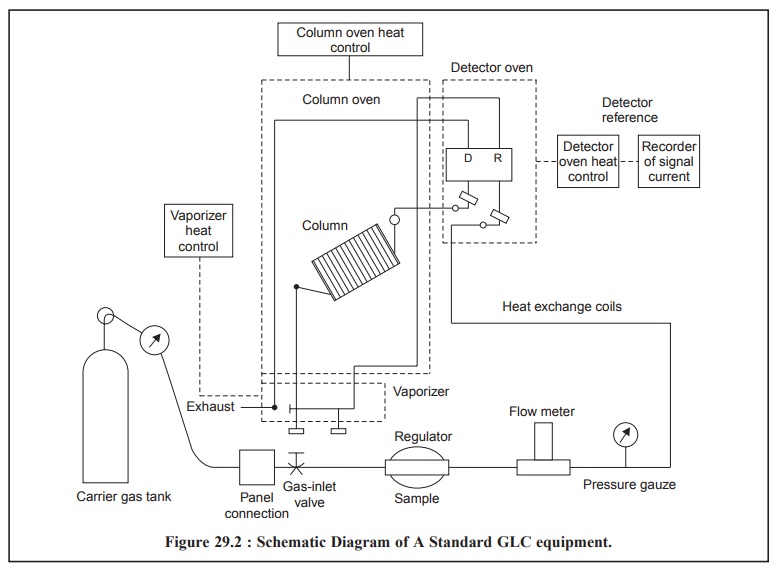
INSTRUMENTATION
A gas chromatograph essentially comprises of six vital components, namely :
(a) Carrier Gas
Regulator and Flow Meter,
(b) Sample
Injection System,
(c) Separation
Column,
(d) Thermal
Compartment,
(e) Detectors,
(f) Recording
of Signal Current, and
(g) Integrator.
These components shall be discussed briefly in the
sections that follow : Figure 29.2, gives the sche-matic diagram of a standard
GLC equipment showing the various parts :
The sample is introduced into the vaporizer and enters
the column along with the carrier gas at a constant flow through the detector
oven. The reference sample also passes through the detector oven into the
column which is maintained by column-oven heat control device. The detector
picks up the signals of the sample as well as the reference substance one after
the other which is duly amplified and the signal current recorded on a
strip-chart recording device or other suitable means. After passing through the
detector oven the vapours of the sample plus the carrier gas leaves the
equipment through an exhaust pipe.
Note : Ultrapure N2
for use in flame-ionization devices may be generated by the Serfass Apparatus
available commercially.
1. CARRIER GAS PRESSURE REGULATOR AND FLOW METER
The various carrier gas used in GC along with their
characteristic features are stated below :
H2 : It has a distinctly better thermal
conductivity and lower density. Demerits are its reactivity with unsaturated
compounds and hazardous explosive nature,
He : It has an excellent thermal conductivity, low
density, inertness and it permits greater flow rates. It is highly expensive,
N2 : It offers reduced sensitivity and is
inexpensive, and
Air : It is employed only when the atmospheric O2
is beneficial to the detector separation.
Importantly, the operating efficiency of a chromatograph
is directly dependent on the maintenance of a highly constant carrier
gas-flow-rate. Carrier gas passes from the tank through a toggle value, a flow
meter, a few feet of metal capillary restrictors, and a 0-4 m pressure gauze.
The flow rate could be adjusted by means of a needle value mounted on the base
of the flow meter and is controlled by the capillary restrictors. On the
downstream side of the pressure regulator, a tee (T) may split the flow and
direct it to the sample and the reference side of the detector.
2. SAMPLE INJECTION SYSTEM
The sample injection system is very important and
critical because GC makes use of very small amounts of the samples. A good and ideal
sample injection system should be the one where the sample must not—
(i) be
decomposed at the point of injection,
(ii) create
pressure surges, and
(iii) undergo
fractionation, condensation or adsorption of components during the course of
transfer to the column.
There are different modes of handling liquid, solid and
gaseous samples in a GC which will be discussed briefly here :
(a)
Liquid Samples : They are usually
injected by hypdermic syringes through a self-sealing silicon-rubber septum
into a preheated-metal-block flash evaporator. The sample is vapourized as a
‘plug’ and carried right into the column by the respective carrier gas. Sample
size ranges between 1–10 μ l.
(b)
Solid Samples : These are either dissolved in
volatile liquids (solvents) or temporarily liquefied by exposure to infra-red
heat.
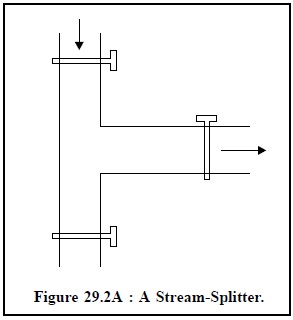
(c) Gas Samples : They are best handled and injected by gas-light syrings or a gas-sampling valve, usually termed as a stream-splitter. In the simplest form this is merely a glass-system (Figure 29.2A) made up of three stop-cocks, between two of which there is a standard volume wherein the ‘gas’ is trapped. Gas from this bypass-capillary-loop is introduced right into the column by sliding or rotating a valve to connect the loop with the stream of carrier gas.
3. SEPARATION COLUMN
It is also known as the ‘chromatographic column’. In reality the heart of a GC is the column
duly packed or capillary in which the separation of constituents is
materialized. The packed-column is usually a tubing having an internal diameter
of 4.0 mm and made up of stainless-steel, copper, cupronickel or glass either
bent in U-shape or coiled. Its length varies from 120 cm to 150 M.
The general requirements of a liquid phas are :
·
Differential partitioning of sample components,
·
Reasonably good solvent properties for components,
·
High thermal stability, and
·
A lower vapour pressure at the column temperature.
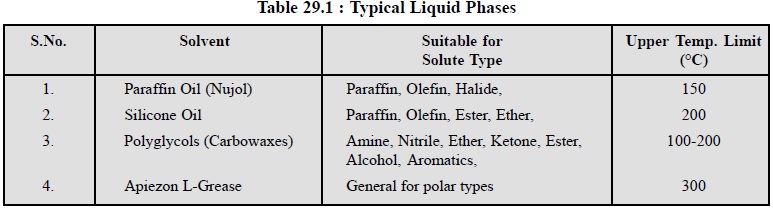
Table 29.1, illustrates the characteristic features of
some typical liquid-phases used in GC :
4. THERMAL COMPARTMENT
A precise control of the column temperature is not only a
must but also a requisite, whether it is intended to maintain an
invariant-temperature or to provide a programmed-temperature. Importantly, the
temperature of the column oven must be controlled by a system that is sensitive
enough to changes of 0.01°C and that main-tains an accurate control to 0.1°C.
In normal practice, an air-bath chamber surrounds the column and air is
circulated by a blower through the thermal compartment. However, separate
temperature controls are very much desirable for the vaporizer block as well as
the detector-oven.
More recently, programmes are also available that
features both in linear and non-linear temperature programming as sample and
reference columns. The compartment temperature can also be raised at various
rates upto a maximum of 60 °C min–1 in the lower-temperature ranges
and about 35 °C min–1 at higher temperatures.
5. DETECTORS
There are in all six
different kinds of detectors used in ‘Gas Chromatography’, namely :
(i) Thermal
conductivity detector (TCD),
(ii) Flame
ionization detector (FID),
(iii) Electron
capture detector (ECD),
(iv) Thermionic
detector (NP–FID)
(v) Flame
photometric detector (FPD), and
(vi)
Photoionization detector (PID).
The first three
detectors are invariably used in GLC and shall be discussed in details below ;
whereas a passing reference shall be made with respect to the second three detectors.
5.1. Thermal Conductivity Detector (TCD)
The thermal conductivity
detector,
or katharometer, was the first ever
detector employed for GLC; and is
still being used today be virtue of its versatility,
stability, simplicity and above all the low-cost.
Principle : The underlying principle of
TCD is that the ability of a gas to dissipate heat, i.e., its thermal conductivity, from a heated body shall change with the composition
of the gas. It may be further explained by the fact that each specific carrier
gas shall have a characteristic thermal conductivity that is picked up
first-and-foremost by the equilibrium temperature of the detecting element to
afford a baseline signal. Evidently, the thermal conductivity of the mixture of
carrier gas plus sample must be altogether different from that of pure carrier
gas ; and while the mixture takes its course through the detector, an obvious
change in the temperature of the detecting element is duly recorded as a
signal.
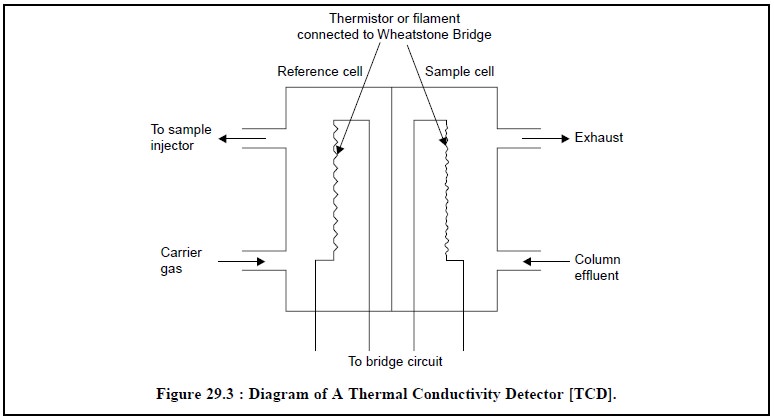
Figure 29.3 shows a simple diagram of a thermal
conductivity detector. It essentially consists of two cells of small volumes,
made within a metal block, termed as reference
cell and sample cell. Each cell
has a resistance wire or thermister or filament that possesses a high
temperature coefficient or resistance i.e.,
the resistance varies appreciably with slight variation in temperature. These
two resistances, namely : reference cell (R) and sample cell (S) are included
in two arms of a Wheatstone Bridge. Now, the carrier gas is passed into both
the cells, but the column-effluents are allowed to enter only the sample cell.
Thus, the temperature of the sample cell changes due to widely different
thermal conductivity of the sample component than that of the carrier gas,
thereby causing a change in resistance of (S) and the Wheatstone Bridge gets
unbalanced. The off-balance current is transmitted to the recorder that finally
draws the elution-curve for the sample(s) undergoing chromatographic
separations.
Cautions
(i) First turn the carrier gas on and then switch on
filament-current/detector block heater, and
(ii) Do not off the carrier gas before switching off the detector
current or before the detector block has attained ambient temperature. This
saves the filament from being damaged and enhances its life-span considerably.
5.2. Flame Ionization Detector (FID)
The general class of ‘ionization detectors’ comprise of the following important
detectors, namely :
·
Flame ionization detector,
·
Electron capture detector,
·
Thermionic detector, and
·
Photoionization detector.
No other detector till date has surpassed the flame
ionization detector (FID) as a universal gas chroma-tographic detector. It
hardly meets, all the characteristic features of TCD in terms of simplicity,
stability, and versatility besides having two
distinctly positive plus points :
(i) Its
linearity over a wider concentration range, and
(ii) It being
more sensitive with less flow and temperature dependency.
Principle : First, the principles of
operation for all ionization detectors shall be discussed briefly and then the actual principles with
specific details would be described under each particular detector.
Generally, the fundamental physical process underlying
the operation of an ionization detector is the conduction of electricity by
gases. At normal temperatures and pressures a gas essentially behaves as a
perfect electrical insulator. However, if electrically charged particles (ions
and electrons) are produced in a gas, it becomes a conductor. In other words,
their free motion in the direction of the electrical field renders the gas
conducting. Assuming a situation, when a vapour is held between two electrodes
to which a voltage is applied, practically and absolutely no current shall flow
at all in the electrical circuit until and unless charged particles are
introduced. The quantum of electric current thus generated would become the
signal of the ionization detector. On applying adequate voltage to the
electrodes, all of the ions would be collected, and hence the ion-current shall
be directly proportional to the number of ions between the electrodes. As the
presence of only a few ions are capable of exhibiting the conductivity of the
gas; therefore, ionization detectors are usually very sensitive.
Principle of FID : The underlying principle of
FID is that invariably a mixture of hydrogen-oxygen or hydrogen-air flame burns with the generation of comparatively
fewer ions, but when an organic compound viz.,
most pharmaceutical substances is ignited in such a flame, ion production gets
enhanced dramatically. There-fore, when such a flame is held between two
electrodes to which a voltage ranging between 100-300 V is applied, it would
instantly give rise to an ion current on burning an organic compound in the
flame.
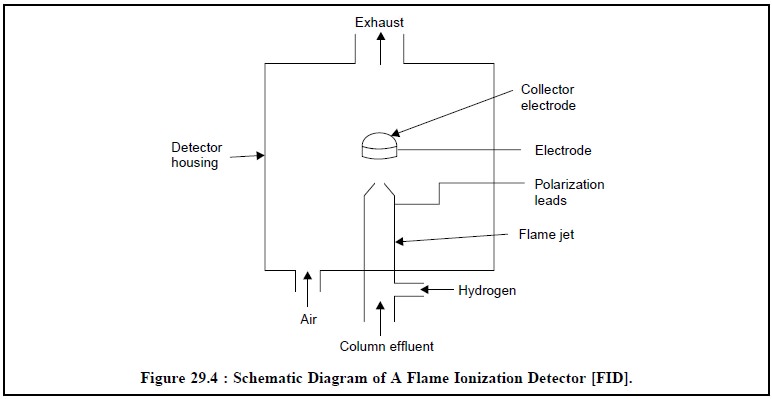
Figure 29.4, illustrates a schematic diagram of a flame-ionization detector. It comprises
of a posi-tively charged ring (also referred to as cylindrical collector
electrode), whereas the flame jet serves as the negative electrode. The flame
jet has two inlets ; from the bottom of the column effluent is introduced and
from the side H2 to form the fuel, whereas air is let in uniformly
around the base of the jet.
5.3. Electron Capture Detector (ECD)
In the domain of gas chromatography the electron capture detector (ECD) enjoys
the reputation of being one of the most
sensitive as well as selective
detectors. However, this valuable detector needs to be handled with a lot
of skill and expertise so as to achieve wonderful and dependable results.
Principle : ECD belongs to the general
class of ionization detectors, the underlying principles of which have already been discussed. In ECD
specifically a β-emitter serves as a source of radiation to generate the
ions that helps in ionizing the carrier gas molecules to form positive ions and
free electrons as expressed in the following Equation (e) :
C + radiation → C+ + e– ...(e)
In a situation when the said phenomenon is conducted
between a pair of charged electrodes, the mobility of the lighter negative ions
i.e., the electrons, would be much
higher in comparison to the heavier positive ions
i.e., the charged
carrier-gas-molecules, thereby ruling out the possibility of their
‘recombination’. Thus mostly the
cations and electrons will be collected, while generating a standing current
that forms the baseline-signal of the ECD detector. At this stage, if an
organic molecule, (i.e., a
pharmaceutical substance) possessing a com-paratively high electron affinity is
introduced, a portion of the electrons shall be captured to produce negatively
charged ions. These heavy-negative-ions will have less mobility as compared to
the electrons ; therefore, they will have no other coice than to unite with
positive ions. Thus, the net result would be fewer ions and electrons available
to migrate to the electrodes, thereby causing a marked and pronounced reduction
in the standing current of the detector. Ultimately, this observed current
decrease represent as the ‘signal’ of the electron capture detector.
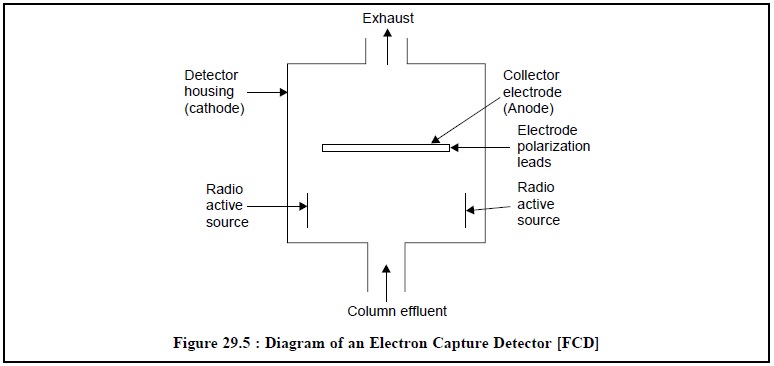
Figure 29.5, depicts the diagram of an electron capture
detector. The metal block of the detector housing itself serves as a cathode,
whereas an electrode polarizing lead suitably positioned in the centre of the
detector housing caters for a collector electrode (anode). The radioactive
source from a beta-emitter is introduced from either sides of the detector housing
below the electrode polarizing lead.
The column-effluent is passed into the detector from the
bottom whereas its exhaust goes out from the top.
5.4. Thermionic Detector (NP-FID)
The very name suggests, the thermionic detector functions
on the principle of ion-current generated by the thermal production of ions. It
may also be invariably termed as a nitrogen
detector, a sulphur detector, a phophorus detector, and a halogen detector by virtue of the fact
that its specificity in detecting organic compounds essentially containing
these elements. Furthermore, it is also widely known as NP-FID because it is
invariably employed for carrying out the analysis
of N- or P-containing organic compounds.
5.5. Flame Photometric Detector (FPD)
Brody and Chaney* in 1966, were the first and foremost to
describe the flame photometric detector
(FPD) which unfortunately could not get enough recognition in the field of gas
chromatographic analysis due to the following reasons, namely :
(i) Its
selectivity, and
(ii) Its poor
commercial availability.
It solely operates on the principle of photon emission.
If P- or S-containing hydrocarbons are ignited in a hydrogen-rich flame, it
gives rise to chemiluminescent species
spontaneously which may subsequently be detected by a suitably
photomultiplier device. Hence, FPD is regarded as a specific detector for P- or
S-containing compounds.
5.6. Photoionization Detector (PID)
Lovelock** in 1960, first introduced the photoionization
detector but unfortunately its reported usages have been more or less scarce.
PID belongs to the generic class of ionization detectors
whose principles have already been discussed earlier. As the very name
signifies the PID induces ionization via
photons emitted by an UV-lamp. A PID detector makes use of a photon energy of
10.2 electron volts (eV) emitted as a Lyman
alpha line***. Only such compounds having ionization potentials less than
10.2 eV shall absorb the UV-radiation and be subse-quently converted to
positive ions. Two-charged electrodes serve as an electric field in the
detector, the cathode becoming the collector electrode for the ions. The
ion-current thus generated, that will be directly proportional to the ion
concentration, then becomes the signal of the detector.
6. RECORDING OF SIGNAL CURRENT
In general, the signal from a gas chromatograph is
recorded continuously as a function of time by means of a potentiometric
device. Most frequently, a recorder of 1-10 mV full-scale deflection (~
10 inches) and having a response time 1 second or less is quite adequate.
![]()
Variable chart speeds between the range of 5-50 mm. min–1
are most preferable in GC.
Essentially in a potentiometric recorder, the input
signal is balanced continuously by a feedback signal making use of a
servomechanism ; whereby a pen strategically connected to this system moves
proportionally along the width of the chart paper, thus recording the signal,
whereas simultaneously the chart paper keeps moving at a constant speed along
its length.
The following important points should be noted before
operating a recorder, namely :
(i) Its ‘zero’
must be adjusted (or synchronized) with the ‘input zero’ otherwise the baseline
might shift with alterations in attenuation of the signal,
(ii) The
amplifier gain must also be adjusted duly so as to avoid completely the
dead-base and oscilla-tion,
(iii) A
recorder with inadequate shielding from AC circuits would display shifting of
its zero point, and
(iv) A
reasonably good recorder having quality performance must be employed so as to
achieve correct recording of analog-signal, a topmost priority towards
quantitative accuracy and precision.
7. INTEGRATOR
An ‘intergrator’
may be regarded as a device that essentially facilitates simultaneous
measurement of areas under the chromatographic peaks in the chromatogram either
by mechanical or electronic means. It is, however, pertinent to mention here that ‘manual techniques’ for determining peak
areas are known, such as :
‘triangulation’, cutting and weighting of peaks, planimetry, but all these methods
are quite time consuming, tedious
and not accurate. Hence, based on the actual need, incorporation of an
appropriate integrator in a reasonably good GC-set up is an absolute necessity.
There are two
types of integrators generally employed in GC, namely :
(a) Ball and Disk Integrator : This is
nothing but a purely mechanical device and installed at one end of the very
strip-chart recorder itself. It carries a pen that writes along a span of about
one inch, reserved for integrator on the recorder chart paper at the end. The
zero line of the integrator moves almost parallel to the base line of the
chromatogram and as soon as a peak appears on the recorder, the integrator-pen
starts moving from right to left the vice-versa
within its one-inch strip. Each one-inch traverse (counted along projection
parallel to signal axis) is usually assigned a value of 100 counts ; the total
number of counts corresponding to a peak are directly proportional to the area
of the peak.
The type of mechanical integrator* affords fairly good
accuracy and precision ; and above all it is quite cheap.
(b) Electronic Integrator : An ‘electronic integrator’ is definitely a
much superior, accurate and dependable device wherein the GC-signal is
converted to a frequency pulse that are accumulated corresponding to a peak and
later on digitally printed out as a measure of the peak area. The main
advantages of an electronic integrator are, namely :
(i) Provides a
much wider linear range,
(ii) Changing
the ‘attenuation’ is not required, and
(iii) Offers
highest precision in peak-area measurement.
Of course, the electronic integrators are quite expensive
Precision of the TWO methods :
The
‘electronic integrator’ is almost 3
times** more accurate and precise
than the ‘ball and disc integrator’
:

GC-Computer System : Nowadays, a large number of
data-processing-computer-aided instruments
for the automatic calculation of various peak parameters, for instance :
relative retention, composition, peak areas etc., can be conveniently coupled
with GC-systems. A commercially available*** fairly sophisticated computer
system of such type are available abundantly that may be capable of undertaking
load upto 100 gas-chromatographs with ample data-storage facilities. In fact,
the installation such as ‘multi GC-systems’ in the routine
analysis in oil-refineries and bulk pharmaceutical industries, and chemical
based industries have tre-mendously cut-down their operating cost of analysis
to a bare minimum.
Related Topics