Chapter: Genetics and Molecular Biology: DNA Synthesis
Error and Damage Correction - Enzymology
Error and Damage Correction
The bacterial DNA polymerases I and III, but not
the eukaryotic DNA polymerases, as they are commonly purified, possess the
ability to correct mistakes immediately upon misincorporation of a nucleoside
triphosphate. The subunit of the eukaryotic DNA polymerases required for this
3’-to-5’ exonuclease activity must be similar to the prokaryotic error
correcting unit, but is less tightly bound to the polymerizing subunit. We know
this because adding the bacterial protein to the eukaryotic polymerase confers
an error correcting 3’-to-5’ exonuclease activity to the eukaryotic enzyme.
Cells also possess the ability to correct
replication mistakes that have eluded the editing activity of the polymerases.
Enzymes recognize mispaired bases and excise them. The gap thus produced is
filled in by DNA polymerase I, or in eukaryotic cells by DNA polymerase β, and sealed with DNA ligase. At first glance, such
a repair mechanism would not seem to have much value. Half the time it would
correct the nucleotide from the incorrect strand. How can the cell restrict its
repair to the newly synthesized strand? The answer is that DNA repair enzymes
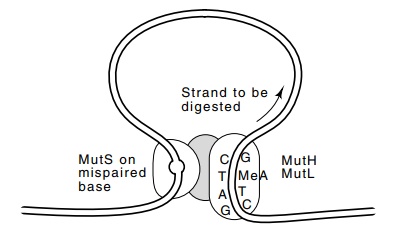
Figure
3.8 The interaction of MutS at a
mispaired base with MutH and MutLactivates resynthesis of the newly synthesized
daughter strand from the GATC sequence past the mispaired base.
distinguish old from new DNA strands with methyl
groups. Only after a newly synthesized strand has been around for some time
does it become methylated and thereby identified as “old.” By this means the
repair enzymes can tell which of the strands should be repaired. Since the
methyl groups involved are not densely spaced along the DNA, the repair enzymes
find the mispaired bases by binding at appropriate sites and moving along the
DNA to the mispaired base, all the while keeping track of which strand is
parental and which is the newly synthesized daughter that should be corrected.
In Escherichia coli, the ages of
strands are identified by the attachment of methyl groups to the ade-nines of
the sequence GATC.
At least one class of mismatched bases that may
exist in Escherichiacoli following
DNA replication is detected by the MutS protein. Thisprotein binds directly to
the mismatched base (Fig. 3.8). Two other proteins of the Mut system, MutH and
MutL, bind to the hemimethy-lated GATC sequence. Apparently, MutH, MutL, and
MutS then interact, generating a nick at the GATC site on the unmethylated
strand. The unmethylated daughter strand is digested from this point and
resynthe-sized by nick translation past the mispaired base, thereby correctly
repairing the original mismatch.
The information stored in the DNA can also be
compromised by damage that occurs subsequent to synthesis. For example, DNA can
be alkylated by chemicals in the environment. As a number of positions in DNA
may be alkylated, a number of proteins exist for removing these groups. This
class of repair system was discovered by the observation that prior exposure of
cells to low concentrations of the mutagen nitrosoguanidine, which alkylates
DNA, greatly reduced the lethality and mutagenicity caused by subsequent
exposure to higher concentra-tions of the agent. This shows that resistance was
induced by the first exposure. Furthermore, the resistance could not be induced
in the presence of protein synthesis inhibitors, also showing that synthesis of
a protein had to be induced to provide resistance.
Additional investigation of the alkylation repair
system has shown that E. coli normally contains about 20
molecules of a protein that can remove methyl groups from O6-methylguanine.
The protein transfers the methyl group from DNA to itself, thereby committing
suicide, and at the same time generating a conformational change so that the
protein induces more synthesis of itself. The methylated protein acts as a
transcription activator of the gene coding for itself. Another domain of this
same protein can transfer methyl groups from methyl phosphotri-ester groups on
the DNA. Additional proteins exist for removal of other adducts.
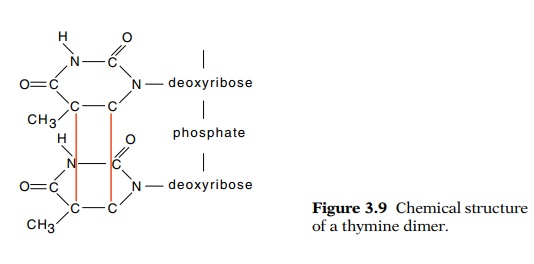
Ultraviolet light can also damage DNA. One of the
major chemical products of UV irradiation is cyclobutane pyrimidine dimers
formed between adjacent pyrimidines, for example between thymine and thymine as
shown in Fig. 3.9. These structures inhibit transcription and replication and must
be removed. The UvrA, UvrB, and UvrC gene products perform this task in E. coli.
In humans, defects in analogs of these enzymes lead to a condition called
Xeroderma pigmentosum, which produces unusual sensitivity to sunlight.
The repair process for UV damaged DNA requires
making nicks in the damaged DNA strand flanking the lesion, removing the
damaged section, and repair synthesis to fill in the gap. Exposure to UV has
long been known to generate mutations. Although the repair process itself could
be inaccurate and generate the mutations, the actual situation is a bit more
complicated. It appears that the existence of the damaged DNA turns off at
least one of the normal error correction pathways. Thus mutations accumulate,
even in portions of chromosomes not damaged by irradiation, if somewhere in the
cell there exists damaged DNA. Ordinarily, pol III utilizes its 3’-5’
exonuclease activity to correct mis-paired bases, but RecA protein can turn off
this repair activity. Appar - ently, RecA protein binds to a damaged site in
the DNA, and as replication passes the point, the protein binds to the
polymerase. The bound RecA then inhibits the normal 3’-5’ exonuclease activity
of the enzyme. Perhaps the relaxation of the fidelity of pol III helps the
enzyme replicate past sites of UV damage. On the other hand, it is possible
that the cell merely utilizes the opportunity of excessive DNA damage to
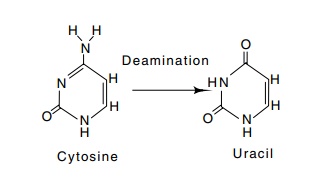
Figure
3.10 Deamination of cytosine produces
uracil.
increase its spontaneous mutation rate. Such a
strategy of increasing mutation rates during times of stress would be of great
evolutionary value. Recent experiments suggest that similar increased mutation
frequencies occur during times of nutrient starvation.
Simple chemical degradation can also compromise the
information stored in DNA. The amino group on cytosine is not absolutely
stable. Consequently, it can spontaneously deaminate to leave uracil (Fig.
3.10). DNA replication past such a point would then convert the former G-C base
pair to an A-T base pair in one of the daughters because uracil has
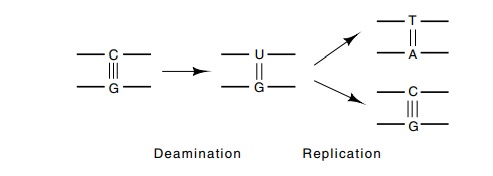
the base-pairing properties of thymine. Enzymes
exist to repair deami-nated cytosine. The first enzyme of the pathway is a
uracil-DNA glycosi-dase that removes the uracil base, leaving the deoxyribose
and phosphodiester backbone. Then the deoxyribose is removed and the resulting
gap in the phosphodiester backbone is filled in. The fact that deaminated
cytosine can be recognized as alien may be the reason thymine rather than
uracil is used in DNA. If uracil were a natural component, cytosine
deaminations could not be detected and corrected.
An interesting example demonstrates the efficiency
of the deami-nated cytosine repair system. As mentioned above, some cells
methylate bases found in particular sequences. In E. coli one such sequence
is CCAGG, which is methylated on the second C. Should this cytosine deaminate,
its extra methyl group blocks the action of the uracil-DNA glycosidase. As a
result, spontaneous deamination at this position cannot be repaired. Indeed,
this nucleotide has been found to be at least ten times as susceptible to
spontaneous mutation as adjacent cytosine residues.
Related Topics