Chapter: Basic & Clinical Pharmacology : Drug Biotransformation
Clinical Relevance of Drug Metabolism
CLINICAL RELEVANCE OF DRUG
METABOLISM
The
dose and frequency of administration required to achieve effective therapeutic
blood and tissue levels vary in different patients because of individual
differences in drug distribution and rates of drug metabolism and elimination.
These differences are determined by genetic factors and nongenetic variables,
such as age, sex, liver size, liver function, circadian rhythm, body
tem-perature, and nutritional and environmental factors such as con-comitant
exposure to inducers or inhibitors of drug metabolism. The discussion that
follows summarizes the most important of these variables.
Individual Differences
Individual
differences in metabolic rate depend on the nature of the drug itself. Thus,
within the same population, steady-state plasma levels may reflect a 30-fold
variation in the metabolism of one drug and only a two-fold variation in the
metabolism of another.
Genetic Factors
Genetic
factors that influence enzyme levels account for some of these differences,
giving rise to “genetic polymorphisms” in drug metabolism. The first examples
of drugs found to be subject to genetic polymorphisms were the muscle relaxant
succinylcholine, the anti-tuberculosis drug isoniazid, and the anticoagulant
warfarin. A true genetic polymorphism is defined as the occurrence of a variant
allele of a gene at a population frequency of ≥ 1%, result-ing in altered expression or
functional activity of the gene product, or both. Well-defined and clinically
relevant genetic polymor-phisms in both phase I and phase II drug-metabolizing
enzymes exist that result in altered efficacy of drug therapy or adverse drug
reactions (ADRs). The latter
frequently necessitate dose adjust-ment (Table 4–4), a consideration
particularly crucial for drugs with low therapeutic indices.
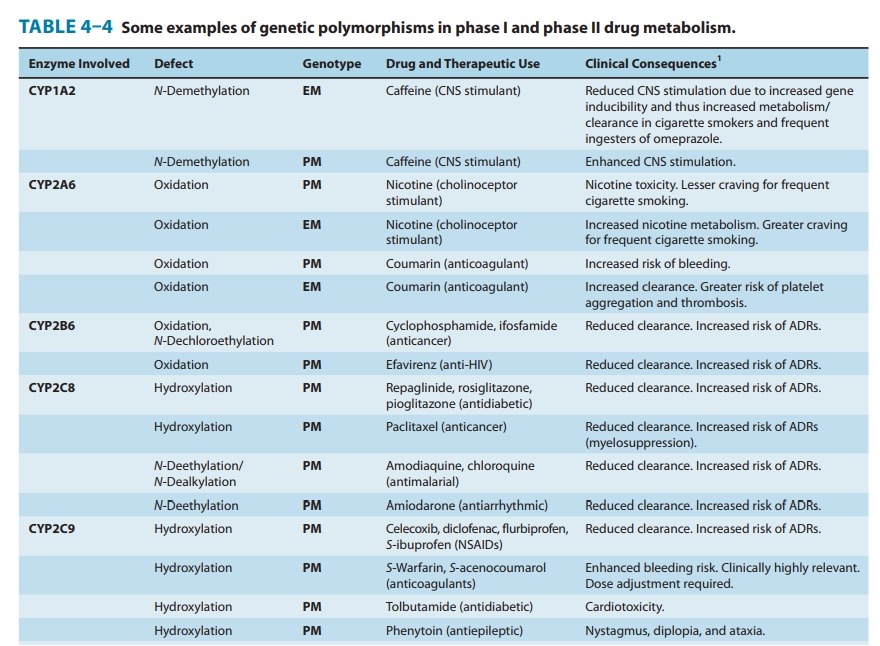
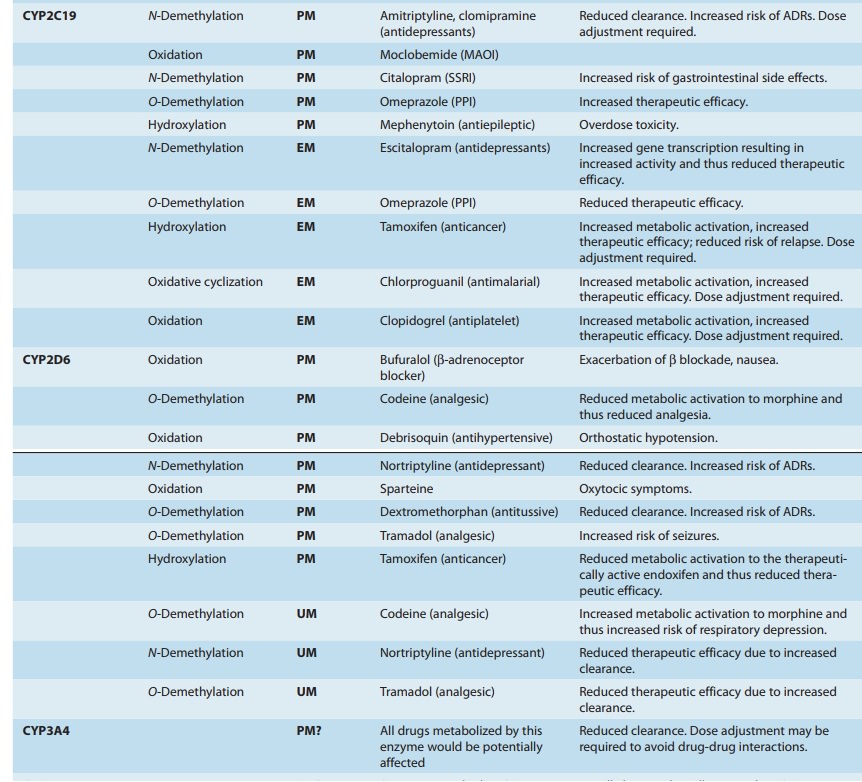
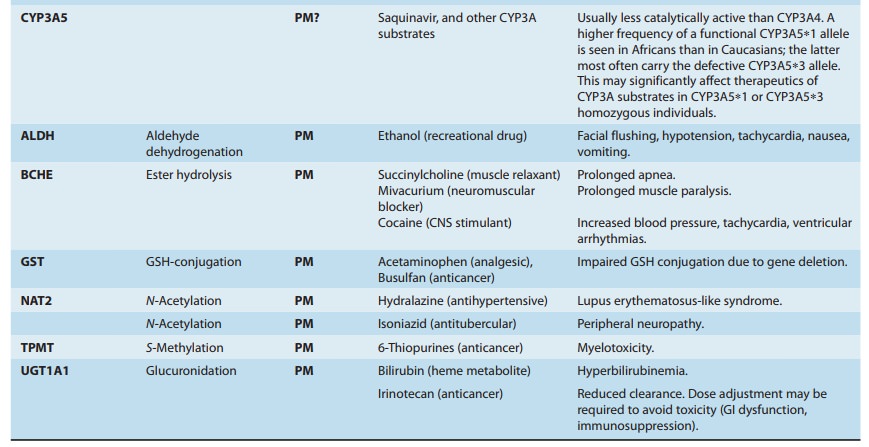
A. Phase I Enzyme Polymorphisms
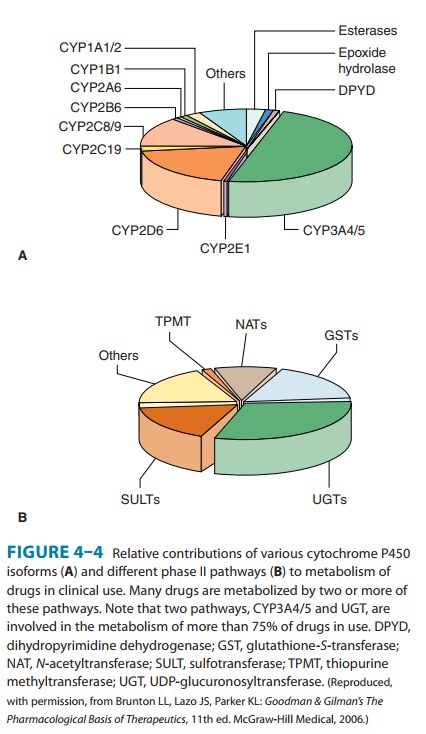
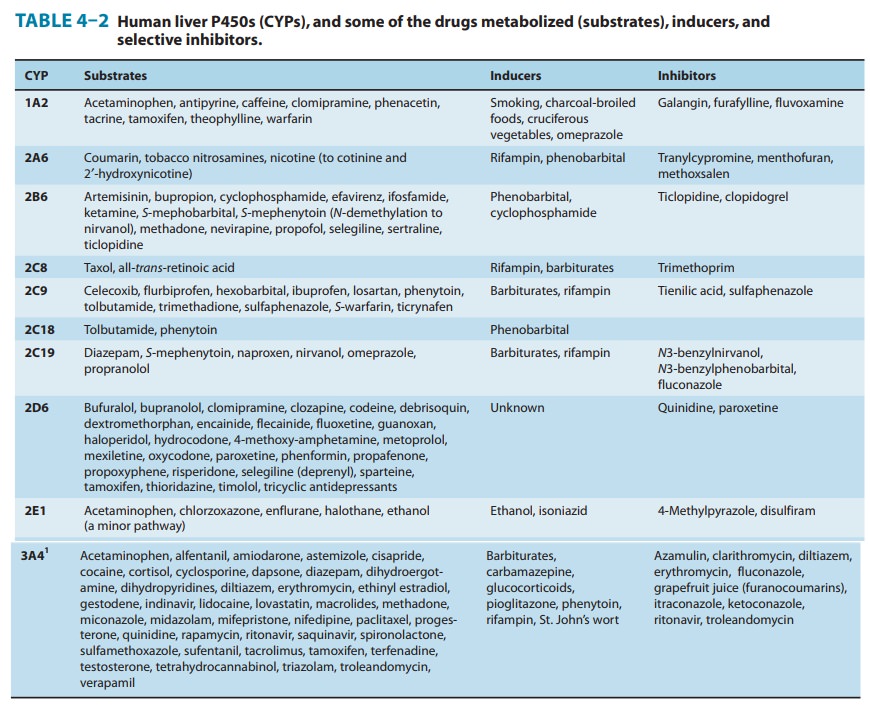
Genetically determined defects in the phase I oxidative metabo-lism of several drugs have been reported (Table 4–4). These defects are often transmitted as autosomal recessive traits and may be expressed at any one of the multiple metabolic transformations that a chemical might undergo. Human liver P450s 3A4, 2C9, 2D6, 2C19, 1A2, and 2B6 are responsible for about 75% of all clinically relevant phase I drug metabolism (Figure 4–4), and thus for about 60% of all physiologic drug biotransformation and elimination. Thus, genetic polymorphisms of these enzymes, by significantly influencing phase I drug metabolism, can alter their pharmacokinetics and the magnitude or the duration of drug response and associated events.
Three
P450 genetic polymorphisms have been particularly well characterized, affording
some insight into possible underlying molecular mechanisms, and are clinically
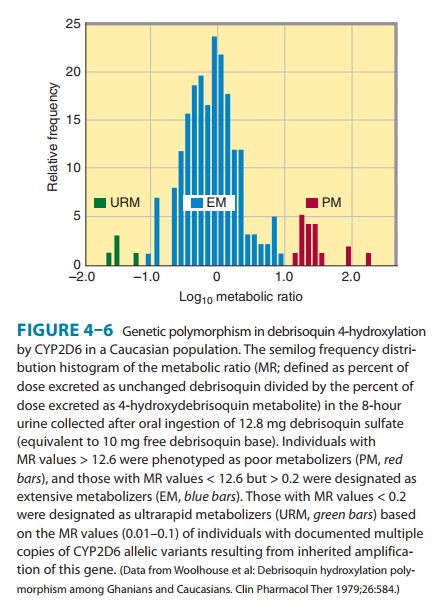
noteworthy, as they require therapeutic dosage adjustment. The first is the debrisoquin-sparteine oxidation type of
polymorphism, which apparentlyoccurs in 3–10% of Caucasians and is inherited as
an autosomal recessive trait. In affected individuals, the CYP2D6-dependent oxidations of debrisoquin and other drugs (Table
4–2; Figure 4–6) are impaired. These defects in oxidative drug metabolism
areprobably co-inherited. The precise molecular basis for the defect appears to
be faulty expression of the P450 protein due to either defective mRNA splicing
or protein folding, resulting in little or no isoform-catalyzed drug metabolism
and thereby conferring a poor
metabolizer (PM) phenotype. This PM phenotype corre-lates with a higher
risk of relapse in patients with breast cancer treated with tamoxifen, an
anti-cancer drug that relies on its CYP2D6-dependent metabolic activation to
endoxifen for its efficacy. More recently, however, another polymorphic
genotype has been reported that results in ultrarapid
metabolism of rele-vant drugs due to the presence of CYP2D6 allelic
variants with
This ultrarapid metabolizer (UM) genotype is most common in Ethiopians and Saudi Arabians,
populations that display it in up to one third of individuals. As a result,
these subjects require two-fold to three-fold higher daily doses of
nortriptyline (an antidepressant and a CYP2D6 substrate) to achieve therapeutic
plasma levels. The poor responsiveness to antidepressant therapy of the UM
phenotype also clinically cor-relates with a higher incidence of suicides
relative to that of deaths due to natural causes in this patient population.
Conversely, in these UM populations the prodrug codeine (another CYP2D6
substrate) is metabolized much faster to morphine, often resulting in
undesirable adverse effects of morphine, such as abdominal pain. Indeed, intake
of high doses of codeine by a mother of the ultrarapid metabolizer type was
held responsible for the morphine-induced death of her breast-fed infant.
The
second well-studied genetic drug polymorphism involves the stereoselective aromatic (4)-hydroxylation of the
anticonvul-sant mephenytoin, catalyzed by CYP2C19.
This polymorphism, which is also inherited as an autosomal recessive trait,
occurs in 3–5% of Caucasians and 18–23% of Japanese populations. It is
genetically independent of the debrisoquin-sparteine polymor-phism. In normal “extensive metabolizers” (EMs) (S)-mephenytoin is extensively
hydroxylated by CYP2C19 at the 4 position of the phenyl ring before its
glucuronidation and rapidexcretion in the urine, whereas (R)-mephenytoin is slowly N-demethylated
to nirvanol, an active metabolite. PMs however,appear to totally lack the
stereospecific (S)-mephenytoin
hydroxy-lase activity, so both (S)-
and (R)-mephenytoin enantiomers are N-demethylated to nirvanol, which
accumulates in much higherconcentrations. Thus, PMs of mephenytoin show signs
of pro-found sedation and ataxia after doses of the drug that are well
tolerated by normal metabolizers. Two defective CYP2C19 variant alleles
(CYP2C19∗2 and CYP2C19∗3), the latter predominant in Asians, are
responsible for the PM genotype. The molecular bases include splicing defects
resulting in a truncated, nonfunctional protein. CYP2C19 is responsible for the
metabolism of various clinically relevant drugs (Table 4–4). Thus, it is
clinically impor-tant to recognize that the safety of each of these drugs may
be severely reduced in persons with the PM phenotype. On the other hand, the PM
phenotype can notably increase the therapeutic efficacy of omeprazole, a
proton-pump inhibitor, in gastric ulcer and gastroesophageal reflux diseases.
Another
CYP2C19 variant allele (CYP2C19∗17)
exists that is associated with increased transcription and thus higher CYP2C19
expression and even higher functional activity than that of the wild type
CYP2C19-carrying EMs. Individuals carrying this CYP2C19∗17 allele exhibit higher metabolic activation
of pro-drugs such as the breast cancer drug tamoxifen, the antimalarial
chlorproguanil, and the antiplatelet drug clopidogrel. The former event is
associated with a lower risk of breast cancer relapse, and the latter event
with an increased risk of bleeding. Carriers of the CYP2C19∗17 allele are also known to enhance the
metabolism and thus the elimination of drugs such as the antidepressants
esci-talopram and imipramine, as well as the antifungal voriconazole. This
consequently impairs the therapeutic efficacy of these drugs, thus requiring
clinical dosage adjustments.
The
third relatively well-characterized genetic polymorphism is that of CYP2C9. Two well-characterized variants
of this enzyme exist, each with amino acid mutations that result in altered
metab-olism. The CYP2C9∗2
allele encodes an Arg144Cys mutation, exhibiting impaired functional
interactions with POR. The other
allelic variant, CYP2C9∗3, encodes an enzyme with an Ile359Leu
mutation that has lowered affinity for many substrates. For example,
individuals displaying the CYP2C9∗3
phenotype have greatly reduced tolerance for the anticoagulant warfarin. The
war-farin clearance in CYP2C9∗3-homozygous
individuals is about 10% of normal values, and these people have a much lower
toler-ance for the drug than those who are homozygous for the normal wild type
allele. These individuals also have a much higher risk of adverse effects with
warfarin (eg, bleeding) and with other CYP2C9 substrates such as phenytoin,
losartan, tolbutamide, and some nonsteroidal anti-inflammatory drugs (Table
4–4).
Allelic
variants of CYP3A4 have also been reported, but their contribution to its
well-known interindividual variability in drug metabolism apparently is
limited. On the other hand, the expres-sion of CYP3A5, another human liver isoform, is markedly poly-morphic,
ranging from 0% to 100% of the total hepatic CYP3A content. This CYP3A5 protein
polymorphism is now known to result from a single nucleotide polymorphism (SNP) within intron 3, which enables
normally spliced CYP3A5 transcripts in 5% of Caucasians, 29% of Japanese, 27%
of Chinese, 30% of Koreans, and 73% of African Americans. Thus, it can
significantly contribute to interindividual differences in the metabolism of
preferential CYP3A5 substrates such as midazolam. Two other CYP3A5 allelic
variants that result in a PM phenotype are also known.
Polymorphisms
in the CYP2A6 gene have also been
recently characterized, and their prevalence is apparently racially linked.
CYP2A6 is responsible for nicotine oxidation, and tobacco smok-ers with low CYP2A6
activity consume less and have a lower incidence of lung cancer. CYP2A6 1B
allelic variants associated with faster rates of nicotine metabolism have been
recently discov-ered. It remains to be determined whether patients with these
faster variants will fall into the converse paradigm of increased smoking
behavior and lung cancer incidence.
Additional
genetic polymorphisms in drug metabolism (eg, CYP2B6) that are inherited independently from those
alreadydescribed are being discovered. For instance, a 20- to 250-fold
variation in interindividual CYP2B6 expression partly due to genetic
polymorphisms has been reported. This may have a sig-nificant impact on the
metabolism of several clinically relevant drugs such as cyclophosphamide,
methadone, efavirenz, selegiline, and propofol. Studies of theophylline
metabolism in monozygotic and dizygotic twins that included pedigree analysis
of various families have revealed that a distinct polymorphism may exist for
this drug and may be inherited as a recessive genetic trait. Genetic drug
metabolism polymorphisms also appear to occur for amin-opyrine and
carbocysteine oxidations. Regularly updated informa-tion on human P450
polymorphisms is available at http://www.imm.ki.se/CYPalleles/.
Although
genetic polymorphisms in drug oxidations often involve specific P450 enzymes,
such genetic variations can also occur in other enzymes. Recently, genetic
polymorphisms in POR, the essential P450 electron donor, have been reported. In
particular, an allelic variant (at a 28% frequency) encoding a POR A503V
mutation has been reported to result in impaired CYP17-dependent sex steroid
synthesis and impaired CYP3A4- and CYP2D6-dependent drug metabolism in vitro.
Its involvement in clinically relevant drug metabolism, while predictable,
remains to be established. Descriptions of a polymorphism in the oxidation of
trimethylamine, believed to be metabolized largely by the flavinmonooxygenase (Ziegler’s enzyme), result in the
“fish-odorsyndrome” in slow metabolizers, thus suggesting that genetic
vari-ants of other non–P450-dependent oxidative enzymes may also contribute to
such polymorphisms.
B. Phase II Enzyme Polymorphisms
Succinylcholine
is metabolized only half as rapidly in persons with genetically determined
deficiency in pseudocholinesterase (now generally referred to as
butyrylcholinesterase [BCHE]) as in
per-sons with normally functioning enzyme. Different mutations, inherited as
autosomal recessive traits, account for the enzyme deficiency. Deficient
individuals treated with succinylcholine as a surgical muscle relaxant may
become susceptible to prolonged respiratory paralysis (succinylcholine apnea).
Similar pharmacoge-netic differences are seen in the acetylation of isoniazid.
The defectin slow acetylators (of isoniazid and similar amines) appears to be
caused by the synthesis of less of the NAT2 enzyme rather than of an abnormal
form of it. Inherited as an autosomal recessive trait, the slow acetylator phenotype occurs in about 50% of blacks and whites
in the USA, more frequently in Europeans living in high northern latitudes, and
much less commonly in Asians and Inuits (Eskimos). The slow acetylator
phenotype is also associated with a higher incidence of isoniazid-induced
peripheral neuritis, drug-induced autoimmune disorders, and bicyclic aromatic
amine-induced bladder cancer.
A
clinically important polymorphism of the TPMT
(thiopurine S-methyltransferase) gene
is encountered in Europeans (frequency,1:300), resulting in a rapidly degraded
mutant enzyme and conse-quently deficient S-methylation
of aromatic and heterocyclic sulfhydryl compounds including the anti-cancer
thiopurine drugs 6-mercaptopurine, thioguanine, and azathioprine, required for
their detoxification. Patients inheriting this polymorphism as an autosomal
recessive trait are at high risk of thiopurine drug-in-duced fatal
hematopoietic toxicity.
Genetic
polymorphisms in the expression of other phase II enzymes (UGTs and GSTs) also
occur. Thus, UGT polymor-phisms (UGT1A1∗28) are associated with
hyperbilirubinemic diseases (Gilbert’s syndrome) as well as toxic side effects
due to impaired drug conjugation and/or elimination (eg, the anticancer drug
irinotecan). Similarly, genetic polymorphisms (GSTM1) in GST (mu1 isoform) expression can lead to significant
adverse effects and toxicities of drugs dependent on its GSH conjugation for
elimination.
C. The Role of Pharmacogenetic Testing in Clinically Safe & Effective Drug Therapy
Despite
our improved understanding of the molecular basis of pharmacogenetic defects in
drug-metabolizing enzymes, their impact on drug therapy and ADRs, and the
availability of vali-dated pharmacogenetic biomarkers to identify patients at
risk, this clinically relevant information has not been effectively translated
to patient care. Thus, the much-heralded potential for personal-ized medicine,
except in a few instances of drugs with a relatively low therapeutic index (eg,
warfarin), has remained largely unreal-ized. This is so even though 98% of US
physicians are apparently aware that such genetic information may significantly
influence therapy. This is partly due to the lack of adequate training in
translating this knowledge to medical practice, and partly due to the logistics
of genetic testing and the issue of cost-effectiveness. Severe ADRs are known
to contribute to 100,000 annual US deaths, about 7% of all hospital admissions,
and an increased aver-age length of hospital stay. Genotype information could
greatly enhance safe and efficacious clinical therapy through dose adjust-ment
or alternative drug therapy, thereby curbing much of the rising ADR incidence
and its associated costs.
Diet & Environmental Factors
Diet
and environmental factors contribute to individual variations in drug
metabolism. Charcoal-broiled foods and cruciferous vegetables are known to
induce CYP1A enzymes, whereas grapefruit juice is known to inhibit the CYP3A
metabolism of co-administered drug substrates (Table 4–2). Cigarette smokers
metabolize some drugs more rapidly than nonsmokers because of enzyme induction
(see previous section). Industrial workers exposed to some pesticides
metabolize certain drugs more rapidly than unexposed individu-als. Such
differences make it difficult to determine effective and safe doses of drugs that
have narrow therapeutic indices.
Age & Sex
Increased
susceptibility to the pharmacologic or toxic activity of drugs has been
reported in very young and very old patients com-pared with young adults.
Although this may reflect differences in absorption, distribution, and
elimina-tion, differences in drug metabolism also play a role. Slower
metabolism could be due to reduced activity of metabolic enzymes or reduced
availability of essential endogenous cofactors.
Sex-dependent
variations in drug metabolism have been well documented in rats but not in
other rodents. Young adult male rats metabolize drugs much faster than mature
female rats or pre-pubertal male rats. These differences in drug metabolism
have been clearly associated with androgenic hormones. Clinical reports suggest
that similar sex-dependent differences in drug metabolism also exist in humans
for ethanol, propranolol, some benzodiaz-epines, estrogens, and salicylates.
Drug-Drug Interactions during Metabolism
Many
substrates, by virtue of their relatively high lipophilicity, are not only
retained at the active site of the enzyme but remain non-specifically bound to
the lipid endoplasmic reticulum membrane. In this state, they may induce
microsomal enzymes, particularly after repeated use. Acutely, depending on the
residual drug levels at the active site, they also may competitively inhibit
metabolism of a simultaneously administered drug.
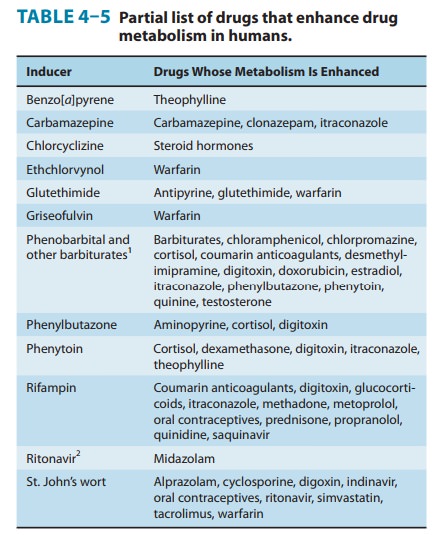
Enzyme-inducing
drugs include various sedative-hypnotics, antipsychotics, anticonvulsants, the
antitubercular drug rifampin, and insecticides (Table 4–5). Patients who
routinely ingest barbi-turates, other sedative-hypnotics, or certain
antipsychotic drugs may require considerably higher doses of warfarin to
maintain a therapeutic effect. On the other hand, discontinuance of the
seda-tive inducer may result in reduced metabolism of the anticoagu-lant and
bleeding—a toxic effect of the ensuing enhanced plasma levels of the
anticoagulant. Similar interactions have been observed in individuals receiving
various combinations of drug regimens such as rifampin, antipsychotics, or
sedatives with contraceptive agents, sedatives with anticonvulsant drugs, and
even alcohol with hypoglycemic drugs (tolbutamide).
It
must also be noted that an inducer may enhance not only the metabolism of other
drugs but also its own metabolism. Thus, continued use of some drugs may result
in a pharmacokinetic type of tolerance—progressively
reduced therapeutic effectiveness due to enhancement of their own metabolism.
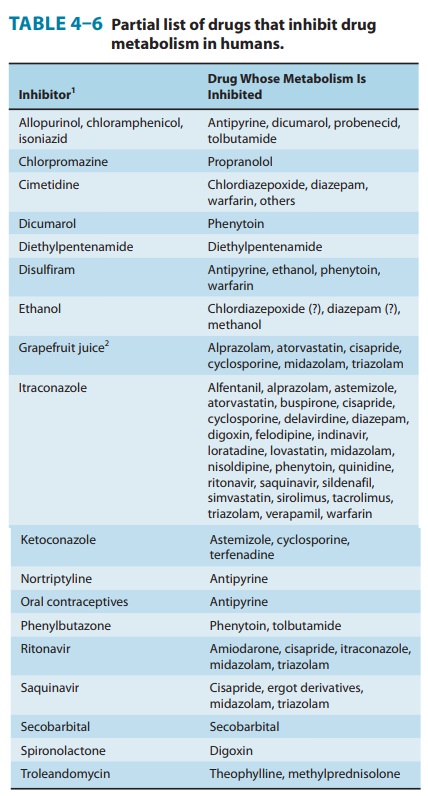
Conversely, simultaneous administration of two or more drugs may result in impaired elimination of the more slowly metabolized drug and prolongation or potentiation of its pharmacologic effects (Table 4–6). Both competitive substrate inhibition and irreversible substrate-mediated enzyme inactivation may augment plasma drug levels and lead to toxic effects from drugs with narrow thera-peutic indices. Indeed, such acute interactions of terfenadine (a second-generation antihistamine) with a CYP3A4 substrate-inhibitor (ketoconazole, erythromycin, or grapefruit juice) resulted in fatal cardiac arrhythmias (torsades de pointe) requiring its with-drawal from the market. Similar drug-drug interactions with CYP3A4 substrate-inhibitors (such as the antibiotics erythromy-cin and clarithromycin, the antidepressant nefazodone, the anti-fungals itraconazole and ketoconazole, and the HIV protease inhibitors indinavir and ritonavir), and consequent cardiotoxicity led to withdrawal or restricted use of the 5-HT4 agonist, cisapride. Similarly, allopurinol both prolongs the duration and enhances the chemotherapeutic and toxic actions of mercaptopurine by competitive inhibition of xanthine oxidase. Consequently, to avoid bone marrow toxicity, the dose of mercaptopurine must be reduced in patients receiving allopurinol. Cimetidine, a drug used in the treatment of peptic ulcer, has been shown to potentiate the pharmacologic actions of anticoagulants and sedatives. The metabolism of the sedative chlordiazepoxide has been shown to be inhibited by 63% after a single dose of cimetidine; such effects are reversed within 48 hours after withdrawal of cimetidine.Impaired metabolism may also result if a simultaneously administered drug irreversibly inactivates a common metabolizing enzyme. These inhibitors, in the course of their metabolism by cytochrome P450, inactivate the enzyme and result in impairment of their own metabolism and that of other cosubstrates. This is indeed the case of the furanocoumarins in grapefruit juice that inactivate CYP3A4 in the intestinal mucosa and consequently enhance its proteolytic degradation.
This impairment of their intes-tinal first-pass
CYP3A4-dependent metabolism significantly enhances the bioavailability of
drugs, such as felodipine, nifedipine, terfenadine, verapamil,
ethinylestradiol, saquinavir, and cyclosporine A, and is associated with
clinically relevant drug-drug and food-drug interactions.Recovery from these
interactions is dependent on CYP3A4 resynthesis and thus may be slow.
Interactions between Drugs & Endogenous Compounds
Some
drugs require conjugation with endogenous substrates such as GSH, glucuronic
acid, or sulfate for their inactivation. Consequently, different drugs may
compete for the same endoge-nous substrates, and the faster-reacting drug may
effectively deplete endogenous substrate levels and impair the metabolism of
the slower-reacting drug. If the latter has a steep dose-response curve or a
narrow margin of safety, potentiation of its therapeutic and toxic effects may
result.
Diseases Affecting Drug Metabolism
Acute
or chronic diseases that affect liver architecture or function markedly affect
hepatic metabolism of some drugs. Such condi-tions include alcoholic hepatitis,
active or inactive alcoholic cirrhosis, hemochromatosis, chronic active
hepatitis, biliary cir-rhosis, and acute viral or drug-induced hepatitis.
Depending on their severity, these conditions may significantly impair hepatic
drug-metabolizing enzymes, particularly microsomal oxidases, and thereby
markedly affect drug elimination. For example, the half-lives of
chlordiazepoxide and diazepam in patients with liver cirrhosis or acute viral
hepatitis are greatly increased, with a cor-responding increase in their
effects. Consequently, these drugs may cause coma in patients with liver
disease when given in ordi-nary doses.Some drugs are metabolized so readily
that even marked reduction in liver function does not significantly prolong
their action. However, cardiac disease, by limiting blood flow to the liver,
may impair disposition of those drugs whose metabolism is flow-limited (Table
4–7). These drugs are so readily metabolized by the liver that hepatic
clearance is essentially equal to liver blood flow. Pulmonary disease may also
affect drug metabolism, as indi-cated by the impaired hydrolysis of
procainamide and procaine in patients with chronic respiratory insufficiency
and the increased half-life of antipyrine (a P450 functional probe) in patients
with lung cancer. The impaired enzyme activity or defective formation of
enzymes associated with heavy metal poisoning or porphyria also results in
reduced hepatic drug metabolism.Although the effects of endocrine dysfunction
on drug metab-olism have been well explored in experimental animal models,
corresponding data for humans with endocrine disorders are scanty. Thyroid
dysfunction has been associated with altered metabolism of some drugs and of
some endogenous compounds as well. Hypothyroidism increases the half-life of
antipyrine, digoxin, methimazole, and some β blockers, whereas hyperthy-roidism has the
opposite effect. A few clinical studies in diabetic patients indicate no
apparent impairment of drug metabolism, although impairment has been noted in
diabetic rats. Malfunctions of the pituitary, adrenal cortex, and gonads
markedly reduce hepatic drug metabolism in rats. On the basis of these
findings, it may be supposed that such disorders could significantly affect
drug metabolism in humans. However, until sufficient evidence is obtained from
clinical studies in patients, such extrapolations must be considered
tentative.Finally, the release of inflammatory mediators, cytokines, and nitric
oxide associated with bacterial or viral infections, cancer, or inflammation
are known to impair drug metabolism by inactivat-ing P450s and enhancing their
degradation.

Related Topics