Chapter: Biotechnology Applying the Genetic Revolution: Bacterial Infections
Cholera Toxin
CHOLERA
TOXIN
The cholera bacterium, Vibrio
cholerae , does not invade the tissues of the host. It attaches to the
exterior of cells lining the small intestine and stays there. The damage is due
to the secretion of cholera toxin. This attacks intestinal epithelial cells, causing
them to lose sodium ions and then water into the intestinal tract. The
clinical symptoms of cholera are loss of
body fluids by massive diarrhea and subsequent death by dehydration.
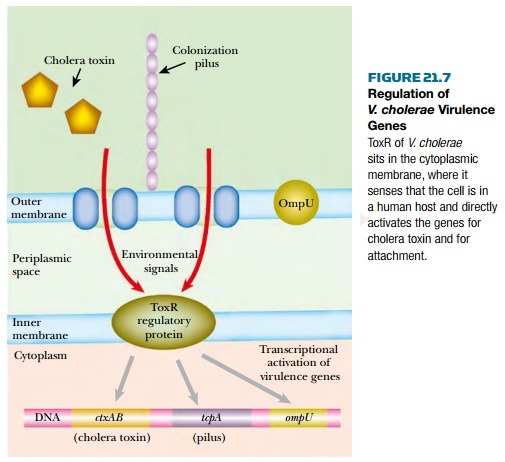
The virulence proteins of Vibrio
cholerae include cholera toxin as well as both pilus-borne and cell-surface
adhesins for binding to intestinal cells. The genes for cholera toxin are carried
by the CTXphi filamentous bacteriophage that lysogenizes Vibrio cholerae. Synthesis
of both adhesins and toxin is co-regulated by the ToxR protein found in the
bacterial inner membrane. This senses whether the bacterium is inside an animal
intestine (temperature, pH, and bile salts are all involved) and activates the
virulence genes accordingly ( Fig. 21.7 ). The internal domain of ToxR protein
binds directly to the promoters of these genes.
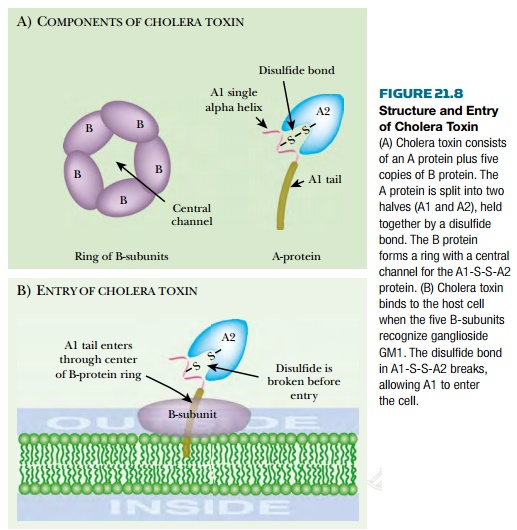
Cholera toxin consists of two proteins, A and B, encoded by the ctxAB genes ( Fig. 21.8 ). The original A protein is split by a protease to give A1 (23.5 kDa) and A2 (5.5 kDa), which remain linked by a single disulfide bond. Five B proteins (11.6 kDa) form a ringlike structure. The A1-S-S-A2 protein protrudes through the ring of five B-subunits. Cholera toxin binds to a glycolipid, known as ganglioside GM1 , in eukaryotic cell membranes. Each B subunit binds to the galactose end of a ganglioside molecule. After binding of the A1A2/B 5 complex to the cytoplasmic membrane, A1 is released from A2 by reduction of the disulfide bond. A1 then enters the cell. Free B protein protects eukaryotic cells against toxin by competing for GM1 binding sites. After entering the intestinal cell, cholera toxin splits NAD into nicotinamide and ADP-ribose, which is then used to ADP-ribosylate target molecules. Cholera toxin A1 protein can actually ADP-ribosylate a variety of acceptors, including free arginine and its derivatives as well as many proteins. A1 protein ADP-ribosylates itself, increasing its activity by 50%. The true in vivolethal target is a G protein involved in regulation of adenylate cyclase ( Fig. 21.9 ).
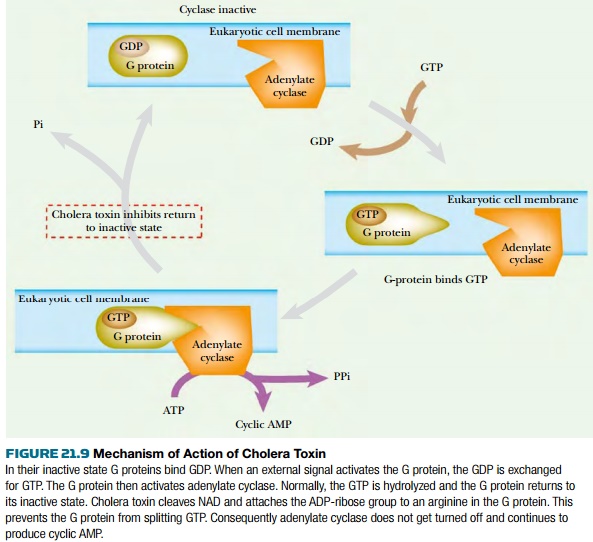
Normally, when the G protein is
activated by a signal from outside the cell, it binds GTP and then binds to and
activates adenylate cyclase. GTP hydrolysis allows the release of the G protein
from the adenylate cyclase and results in deactivation. ADP-ribosylation of an
arginine residue by cholera toxin prevents the hydrolysis of GTP and locks the
G protein in its GTP binding state. This causes hyperactivation of adenylate cyclase
and overproduction of cyclic AMP, which results in the loss of sodium ions and
water. GTP analogs that cannot be hydrolyzed (such as GMP-P-NH-P or GMP-P-CH 2 -P)
show effects similar to cholera toxin.
Cholera toxin and the heat-labile
enterotoxins of other enteric bacteria are actually variants of the same toxin,
although cholera toxin is more potent. The heat-labile enterotoxins found in
some pathogenic strains of E. coli are encoded on the Ent-plasmid, which
may be transferred between strains. These toxins have similar amino acid
sequences to cholera toxin and cause loss of intestinal fluids by the same mechanism.
Related Topics