Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Gene Therapy
Adeno Associated Virus Vectors - Viral Vectors for Gene Transfer
Viral Vectors for Gene Transfer
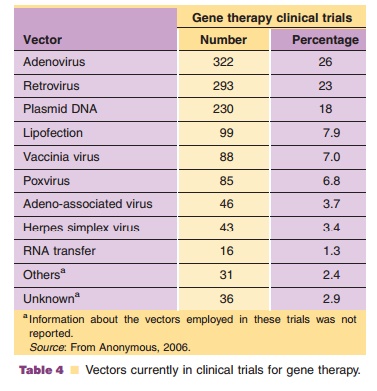
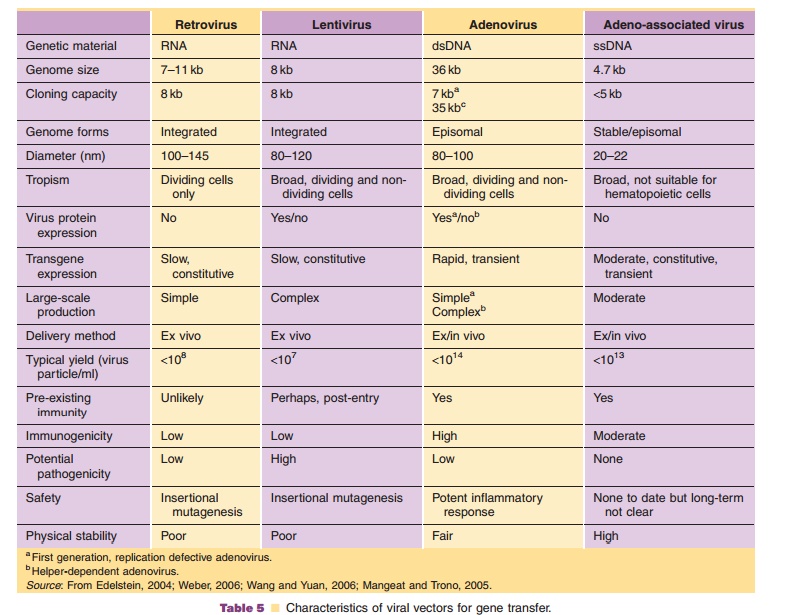
Viruses, natural parasites that efficiently enter cellular targets and hijack cellular machinery for propagation, are currently the most effective vectors for gene therapy. Approximately 70% of all gene therapy clinical trials employ viral vectors (Table 4). Many viruses with divergent properties have been devel-oped for gene transfer. Retroviruses, adenoviruses and adeno-associated viruses (AAVs) are the most extensively studied to date. Characteristics of each are summarized in Table 5.
Adeno-Associated Virus Vectors
Biology
The first human AAV was discovered in 1965 as a contaminant of adenovirus preparations (Atchison, 1965). AAV is a non-enveloped parvovirus that requires co-infection with a helper virus in order to replicate. The AAV genome is a 4.7 kb linear, single stranded DNA molecule composed of two large open reading frames (ORFs), rep and cap, that contain sequences for four replication proteins and three capsid proteins respectively (Fig. 11A). The ORFs are flanked on either side by ITRs, required for genome replication, packa-ging and integration. The left ORF encodes four replication proteins. Rep 68 and 78 play a role in every aspect of the AAV life cycle such as transcription, viral DNA replication and site-specific integration. Rep 40 and 52 are important for DNA packaging in viral capsids within the nucleus. The right ORF contains sequences for structural capsid proteins.
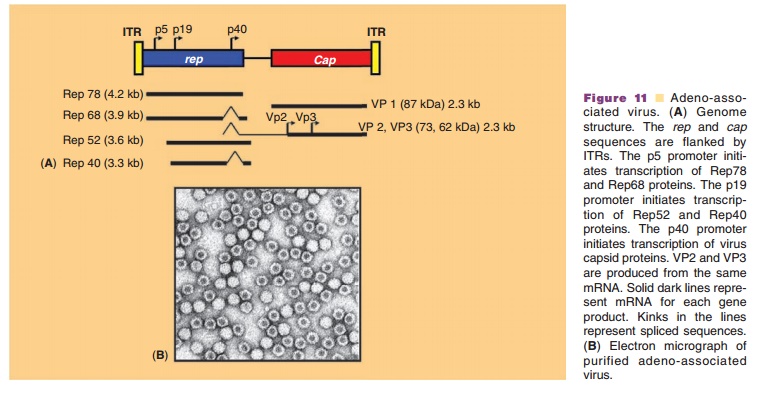
The icosahedral AAV capsid is 25 nm in dia-meter (Fig. 11B). It is relatively simple, made up of only 60 copies of three structural capsid proteins, VP1, VP2, and VP3 in a ratio of 1:1:8 respectively. VP3 interacts with cellular receptors and dictates tissue specificity of the virus. A phospholipase domain, essential for virus infectivity, is located in the N-terminus of VP1. The specific role of VP2 is currently unclear. Recently it was found that viable recombinant AAV vectors could be produced without this protein (Warrington, 2004). To date, 9 distinct AAV serotypes have been identified and over 100 AAV variants have been found in human and non-human primate tissues (Wu, 2006). None of these serotypes cause human disease. The biology of AAV serotype 2 (AAV2) has been the most extensively studied and used as a vector for gene transfer.
AAV2 virions utilize heparan sulfate proteogly-cans (HSPGs) as primary cellular attachment recep-tors, which confer a broad tropism to this virus in skeletal muscle, liver, brain, and retina. (Summerford, 1998). Virus internalization is aided by avb5 integrins (Summerford, 1999), fibroblast growth factor receptor type 1 (FGF-1) (Qing, 1999) and the hepatocyte growth factor receptor, c-Met (Kashiwakura, 2005). The virus enters the cell through receptor-mediated endocytosis and is subsequently transported to the nucleus (Fig. 12). In the nucleus, the single-stranded genome must be converted to a double-stranded form for gene expression. Self-complimentary sequences in the viral ITRs form hairpin structures that serve as origins of replication. Rep 68 and 78 bind to the ITRs and mediate replication. The virus relies upon host cellular machinery as well as helper functions supplied by a coinfecting virus such as adenovirus, herpes simplex virus, or vaccinia, in order to replicate (Muzyczka, 2001). Proteins expressed from the helper virus facilitate transcription, AAV gene expression and DNA replication. Release of virus particles from the host is facilitated by the lysogenic properties of the helper virus. In the absence of helper virus, AAV establishes a latent infection within the cell either by site-specific integration into a unique locus on human chromosome 19, AAVS1, or persisting as a stable
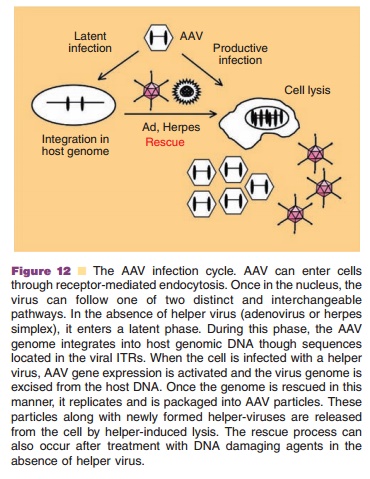
episomal complex (Vasileva, 2005). When the cell is re-infected with helper virus, AAV replication occurs. Chemical carcinogens and ultraviolet radiation can also induce AAV to exit its latent state and replicate. In the context of transgene expression, there is initially a slow rise in transgene levels several weeks after administration of recombinant AAV until a stable plateau is reached. The exact reason for the delay in transgene expression is not clear, although it may reflect requirements for cytoplasmic trafficking, vector uncoating and conversion of the single-stranded genome to the double-stranded form necessary for cellular gene expression.
Suitability of AAVs for Gene Transfer
Recombinant AAV vectors have rapidly gained popularity for gene therapy applications within the last decade, due to their lack of pathogenicity and ability to establish long-term gene expression (Table 5). The viral genome is also simple, making it easy to manipulate. The virus is resistant to many physical and chemical factors, giving AAV a robust stability profile during purification and long-term storage (Croyle, 2001; Wright, 2003). The ability of the virus to integrate in the human chromosome was an initial concern, but it has been found that recombinant AAV vectors that do not express rep proteins integrateat frequencies of approximately 1/3 107 AAV genomes/gene, much lower than the rate of sponta-neous mutations in human genes (1/1 105 muta-tions/gene)(Cole, 1994; Carter, 2005).
Because recombinant AAV vectors do not con-tain any viral ORFs, they do not provoke the innate immune response. The primary host response that affects clinical use of AAV vectors, is production of neutralizing antibodies to capsid proteins, which prevent transgene expression upon readministration (Zaiss, 2005). In addition, approximately 80% of the population is seropositive for antibodies to AAV serotype 2 (Erles, 1999). Cellular and humoral immune responses have also been detected against the transgene product encoded in AAV vectors (Zaiss, 2005; Manno, 2006). These responses depend on the nature of the transgene, the promoter used, the route and site of vector administration, the vector dose and genetic predisposition of the host. Induction of tolerance by adoptive transfer of CD4þCD25þ regulatory T-cells and administration of anti-CD4 and anti-CD40 ligand antibodies, soluble CTLA-4 immunoglobulin fusion proteins or cyclophospha-mide with the first dose of vector have been used to dampen this response. Others have changed the AAV serotype with each dose or created vectors pseudotyped with capsids from different AAV strains (Wu, 2006). In contrast to other vectors, AAV has a limited capacity for transgene cassettes. Because they can join via recombination within ITR regions, the transgene cassette can be split over two vectors given simultaneously although transduction efficiency is reduced by the recombination requirement.
Clinical Use of AAV Vectors
To date, 46 phase I and phase II clinical trials employing recombinant AAV vectors have been initiated (Table 4). Early clinical development of AAV vectors was limited to indications for mono-genetic diseases such as CF, and hemophilia B, but a recent upsurge of trials for other indications was ignited by the impressive safety profile of the virus. The first clinical use of recombinant AAV was to transfer the cystic fibrosis transmembrane-conduc-tance regulator (CFTR) cDNA to the respiratory epithelium for the treatment of CF (Flotte, 1996). These trials were the first to suggest that gene transfer could affect pulmonary function in a positive manner for this disease. Several phase I trials for the treatment of Canavan’s, Parkinson’s and Alzheimer’s disease have shown that AAV-mediated gene transfer to the brain is safe and effective (Carter, 2005; McPhee, 2006). There are currently 4 trials using AAV2 vectors in phase III testing for metastatic hormone-resistant prostate cancer (Simons, 2006).
Production and Processing of Recombinant AAVs
Recombinant AAV vectors are constructed by repla-cing rep and cap genes with a promoter and transgene flanked by viral ITRs. The recombinant virus genome is cloned in a plasmid that is co-transfected in 293 cells with a separate plasmid containing rep and cap without ITRs to prevent packaging of these elements in AAV capsids. Because virus replication requires helper sequences, transfected cells were often infected with a low concentration of adenovirus (Fig. 13A). Early purification protocols capitalized on differences in thermostability and density to effectively separate the two viruses. Adenovirus could be inactivated by placing a preparation at 56˚C for 30 minutes and removed by density gradient ultracentrifugation. Identification of adenoviral sequences that provide helper functions (E1a, E1b, E2a, E4, and VA1) and cloning them into a third plasmid improved the purity of vector stocks (Fig. 13B). Due to the relative inefficiency of triple transfection during large-scale production, cell lines containing integratedrep and cap sequences, the AAV genome or both were developed (Fig. 13C). Toxicity of the rep and cap genes and the need for infection with helper virus continues to complicate AAV production.
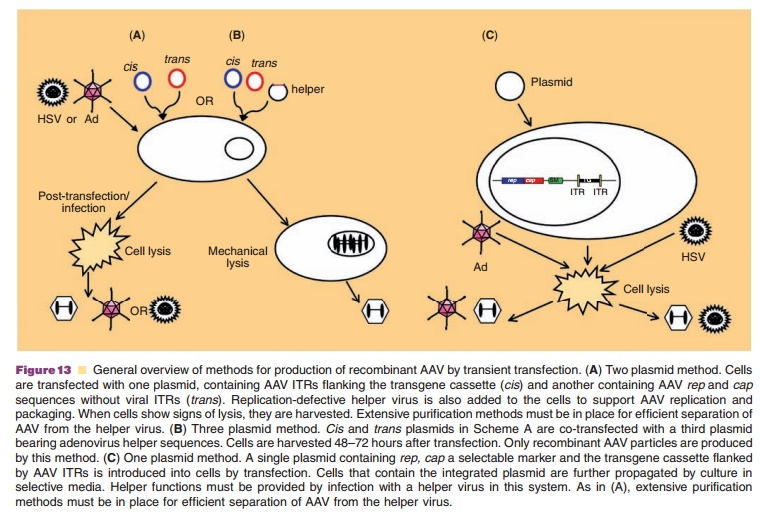
Toxicity of CsCl, aggregation of AAV particles and the fact that adenovirus is not completely removed after extensive centrifugation complicates AAV purification by CsCl density gradient ultracentrifugation (Grieger, 2005). Another density separation medium, iodixanol, that is less toxic than CsCl and prevents AAV aggrega-tion has been employed in a single centrifugation step (Burova, 2005). Passage of the AAV fraction over an affinity column consisting of either a heparinized support matrix or a monoclonal antibodies produced against AAV2 significantly increased purity and in-fectivity of final preparations (Grimm, 1998; Auricchio, 2002). These methods, however, are for specific AAV serotypes. Ion exchange chromatography is the most powerful and versatile method for AAV purification although buffer pH, detergent concentration and column medium must be tailored for each AAV serotype. Infectivity and purity of preparations obtained from these purification strategies are comparable to those obtained from affinity chromatographic methods and are complete within 3 hours.
Few studies have been published that describe systematic efforts to optimize AAV vector formulation and stability. Initial studies demonstrated that AAV is significantly more stable than other recombinant viruses under development for gene transfer. Some have reported that the virus is stable at 4˚C in phosphate buffered saline alone for over 4 months before a significant drop in infectious titer was observed (Croyle, 2001). Similar results have been reported at 25˚C. Addition of cryoprotectants such as sorbitol and polysorbate 80 have extended the stability of the virus at 4˚C for 1 year (Wright, 2003). Lyophilization has also stabilized AAV for over one year at 25˚C after a subtle drop in titer due to processing (Croyle, 2001). Like the adenovirus, AAV is susceptible to loss of infectivity after freezing at –80 and –20˚C. Use of potassium-based buffers that avoid drastic changes in pH during freezing and the addition of cryoprotectants moderately extended the stability of AAV at sub-zero temperatures (Croyle, 2001; Wright, 2003). Aggregation of virus particles and subsequent loss of infectious titer is a significant issue in preparations purified by column chromatography (Wright, 2005). Unlike density gradient ultracentrifu-gation, chromatographic techniques cannot distin-guish between virus capsids that contain the AAV genome and those that are empty, which promote virus aggregation. Increasing the ionic strength of buffers used during purification and processing and in the final formulation significantly reduces this phenomenon and improves long-term stability.
Related Topics