Chapter: Pharmaceutical Biotechnology: Fundamentals and Applications : Monoclonal Antibodies: From Structure to Therapeutic Application
PK/PD in Clinical Development of Antibody Therapeutics
PK/PD in Clinical Development of Antibody Therapeutics
Several new developments have taken place in the antibody therapeutics in the last years. The emphasis in the field has grown and is obvious by the fact that many of the companies are now involved in building antibody product based collaborations. Drug devel-opment has traditionally been performed in sequential phases, divided into preclinical as well as clinical phases I-IV. During the development phases of the molecules, the safety and PK/PD characteristics are established in order to narrow down on the compound selected for development and its dosing regimen. This information-gathering process has recently been char-acterized as two successive learning-confirming cycles (Sheiner, 1997; Sheiner and Wakefield, 1999).
The first cycle (Phases I and IIa) comprises learning about the dose that is tolerated in healthy subjects and confirming that this dose has some measurable benefits in the targeted patients. An affirmative answer at this first cycle provides the justification for a larger and more costly second learn-confirm cycle (Phases IIb and III), where the learning step is focused on how to use the drug benefit/risk ratio, whereas the confirm step is aimed at demon-strating acceptable benefit/risk in a large patient population (Meibohm and Derendorf, 2002). In the following sections we have provided a case study with efalizumab as an approved therapeutic antibody to understand the various steps during the develop-ment of antibodies for various indications.
A summary of the overall PK/PD data from multiple studies within the efalizumab (Raptiva ) clinical development program and an integrated overview of how these data were used for develop-ment and the selection of the approved dosage of efalizumab for psoriasis will be discussed in detail. Psoriasis is a chronic skin disease characterized by abnormal keratinocyte differentiation and hyperpro-liferation and by an aberrant inflammatory process in the dermis and epidermis. T-cell infiltration and activation in the skin and subsequent T-cell-mediated processes have been implicated in the pathogenesis of psoriasis (Krueger, 2002).
Efalizumab is a subcutaneously (SC) adminis-tered recombinant humanized monoclonal IgG1antibody that has received approval for the treatment of patients with psoriasis in more than 30 countries, including the United States and the European Union (Raptiva, 2004). Efalizumab is a targeted inhibitor of T-cell interactions (Werther et al., 1996). An extensive preclinical research program was conducted to study the safety and mechanism of action (MOA) of efalizumab. Multiple clinical studies have also been conducted to investigate the efficacy, safety, PK, PD, and MOA of efalizumab in patients with psoriasis.
Pre-Phase I Studies
In the process of developing therapeutic antibodies integrated understanding of the (PK/PD) concepts provide a highly promising tool. A thorough and rigorous preclinical program in the early learning phase of preclinical drug development can provide a linkage between drug discovery and preclinical development. As it sets the stage for any further development activities, the obtained information at this point is key to subsequent steps (Meibohm and Derendorf, 2002). At the preclinical stage, potential applications might comprise the evaluation of in vivo potency and intrinsic activity, the identification of bio-/surrogate markers, understanding the MOA as well as dosage form/regimen selection and optimiza-tion. A few of these specific aims are described below with information on efalizumab as an example.
Identification of MOA and PD Biomarkers
The identification of appropriate PD endpoints is crucial to the process of drug development. Thus, biomarkers are usually tested early during explora-tory preclinical development for their potential use as PD or surrogate endpoints.
Through an extensive preclinical research pro-gram, the MOA and PD biomarkers for efalizumab have been established. Efalizumab binds to CD11a, the α-subunit of leukocyte function antigen-1 (LFA-1), which is expressed on all leukocytes, and decreases cell surface expression of CD11a. Efalizumab inhibits the binding of LFA-1 to intercellular adhesion molecule-1 (ICAM-1), thereby inhibiting the adhesion of leukocytes to other cell types. Interaction between LFA-1 and ICAM-1 contributes to the initiation and maintenance of multiple processes, including activa-tion of T lymphocytes, adhesion of T-lymphocytes to endothelial cells, and migration of T-lymphocytes to sites of inflammation, including skin. Consistent with the proposed MOA for efalizumab, in vitro experi-ments have demonstrated that efalizumab binds strongly to human lymphocytes with a Kd of approximately 110 ng/mL (Werther et al., 1996; Dedrick et al., 2002) and blocks the interaction of human T-lymphocytes with tissue-specific cells suchas keratinocytes in a concentration-dependent manner.
Upon understanding the MOA, PD effects relevant to the MOA of efalizumab are usually measured in order to identify the efficacious dosage of antibody therapeutics. As saturation of CD11a binding sites by efalizumab has been shown to increase while T-cell activation is increasingly inhib-ited, maximum saturation of CD11a binding sites occurs at efalizumab concentrations >10 µg/mL, re-sulting in maximum T-cell inhibition (Werther et al., 1996; Dedrick et al., 2002). Therefore, CD11a expres-sion and saturation have been chosen as relevant PD markers for this molecule.
Role of Surrogate Molecules
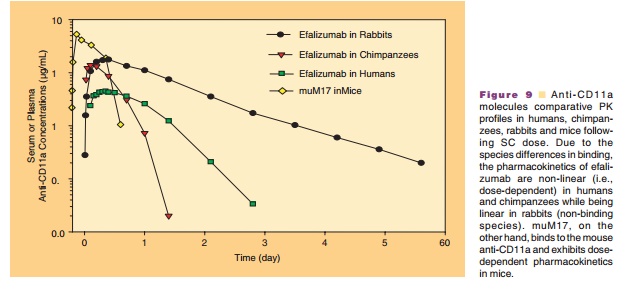
The role of surrogate molecules in assessing ADME of therapeutic antibodies is important as the antigen specificity limits ADME studies of humanized mono-clonal antibodies in rodents. In the development of therapeutic antibodies various molecules may be used to provide a comprehensive view of their PK/PD properties. Studies with surrogates might lead to important information regarding safety, mechanism of action, disposition of the drug, tissue distribution and receptor pharmacology, which might be too cumber-some and expensive to conduct in non-human primates. Surrogates (mouse/rat) provide a means to gaining knowledge of PK and PD in a preclinical rodent model thus allowing rational dose optimiza-tion in the clinic. Therefore in the case of efalizumab to complete a more comprehensive safety assessment, a chimeric rat anti-mouse CD11a antibody, muM17, wasdeveloped and evaluated as a species-specific surro-gate molecule for efalizumab. muM17 binds mouse CD11a with specificity and affinity similar to those of efalizumab to human. In addition, muM17 in mice was demonstrated to have similar pharmacological activities as that of efalizumab in human (Nakakura et al., 1993; Clarke et al., 2004). Representative PK profiles of efalizumab and muM17 in various species are depicted in Figure 9 to help understand the species differences in the PK behavior of molecules.
PK of Efalizumab
A brief overview of efalizumab non-clinical PK/PD results is provided in the following sections to summarize the key observations that led to decisions in designing the subsequent clinical programs. The ADME program consisted of PK, PD (CD11a down-modulation and saturation), and toxicokinetic data from PK, PD, and toxicology studies with efalizumab in chimpanzees and with muM17 in mice. The use of efalizumab in the chimpanzee and muM17 in mice for PK and PD and safety studies was supported by in vitro activity assessments. The non-clinical data were used for PK and PD characterization, PD-based dose selection, and toxicokinetic support for confirm-ing exposure in toxicology studies. Together, these data have supported both the design of the non-clinical program and its relevance to the clinical program.
The observed PD as well as the mechanism of action of efalizumab and muM17 are attributed to their binding to CD11a present on cells and tissues. The binding affinities of efalizumab to human and chimpanzee CD11a on CD3 lymphocytes are compar-able confirming the use of chimpanzees as a valid non-clinical model for humans. CD11a expression has been observed to be greatly reduced on T-lymphocytes in chimpanzees and mice treated with efalizumab and muM17, respectively. Expression of CD11a is restored as efalizumab and muM17 are eliminated from the plasma. The bioavailability of efalizumab in chimpan-zees and muM17 in mice after an SC dose was dose-dependent and ranged from 35% to 48% and 63% to 89% in chimpanzees and mice, respectively. Binding to CD11a serves as a major pathway for clearance of these molecules, which leads to non-linear PK depending on the relative amounts of CD11a and efalizumab or muM17 (Coffey et al., 2005).
The disposition of efalizumab and the mouse surrogate muM17 is mainly determined by the combination of both specific interactions with the ligand-CD11a and by their IgG1 framework and is discussed in detail as follows. The factors control-ling the disposition of these antibodies are shown in Figure 10 and include the following:
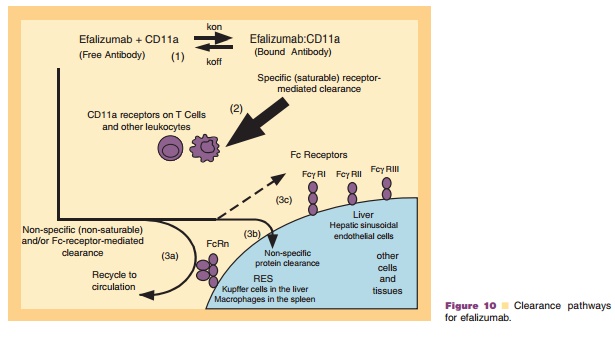
The binding of the free antibody with its ligand CD11a present on both circulating lymphocytes and tissues leads to its removal from circulation. Data suggests that anti-CD11a antibodies are internalized by purified T-cells and upon inter-nalization, the antibodies appeared to be targeted to lysosomes and cleared from within the cells in a time-dependent manner. CD11a-mediated inter-nalization and lysosomal targeting of efalizumabmay constitute one pathway by which this anti-body is cleared in vivo (Coffey et al., 2005).
2. Binding to CD11a is both specific and saturable as demonstrated by the dose dependent clearance of efalizumab in chimpanzees and humans or muM17 in mice.
3. Because of its IgG1 framework, free or unbound efalizumab or muM17 levels are also likely to be influenced by:
a. recycling and circulation following binding to and internalization by the neonatal Fc recep-tor (FcRn),
b. non-specific uptake and clearance by tissues,
c. binding via its Fc framework to Fc receptors present on hepatic sinusoidal endothelial cells.
The disposition of efalizumab is governed by the species specificity and affinity of the antibody for its ligand CD11a, the amount of CD11a in the system, and the administered dose.
Based on the safety studies, efalizumab was generally well tolerated in chimpanzees at doses up to 40 mg/kg/week IV for 6 months, providing an exposure ratio of 339-fold based on cumulative dose and 174-fold based on the cumulative AUC, com-pared with a clinical dose of 1 mg/kg/wk. The surrogate antibody muM17 was also well tolerated in mice at doses up to 30 mg/kg/week SC. In summary efalizumab was considered to have an excellent non-clinical safety profile thereby support-ing the use in adult patients.
Clinical Program of Efalizumab: PK/PD Studies, Assessment of Dose, Route, and Regimen
The drug development process at the clinical stage provides several opportunities for integration of PK/ PD concepts. Clinical Phase I dose escalation studies provide, from a PK/PD standpoint, the unique chance to evaluate the dose-concentration-effect relationship for therapeutic and toxic effects over a wide range of doses up to or even beyond the maximum tolerated dose under controlled conditions (Meredith et al., 1991). PK/PD evaluations at this stage of drug development can provide crucial information regard-ing the potency and tolerability of the drug in vivo and the verification and suitability of the PK/PD concept established during preclinical studies.
Efalizumab PK and PD data are available from 10 studies in which more than 1700 patients with psoriasis received IV or SC efalizumab. In the Phase I studies, PK and PD parameters were characterized by extensive sampling during treatment; in the Phase III trials, steady-state trough levels were measured once or twice during the first 12-week treatment period for all the studies and during extended treatment periods for some studies. Several early Phase I and II trials have examined IV injection of efalizumab and dose-ranging findings from these trials have served as the basis for SC dosing levels used in several subsequent phase I and all Phase III trials.
Administration of Efalizumab
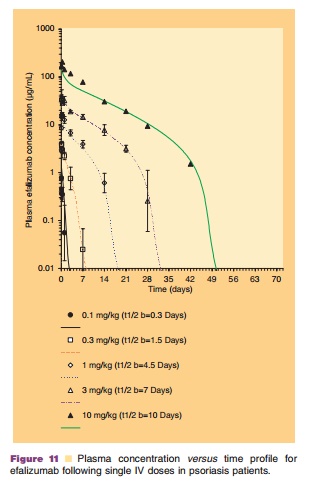
The PK of monoclonal antibodies varies greatly, depending primarily on their affinity for and the distribution of their target antigen (Lobo et al., 2004). Efalizumab exhibits concentration-dependent non-linear PK after administration of single IV doses of 0.03, 0.1, 0.3, 0.6, 1.0, 2.0, 3.0, and 10.0 mg/kg in a Phase I study. This non-linearity is directly related to specific and saturable binding of efalizumab to its cell surface receptor, CD11a, and has been described by a PK/PD model developed by Bauer et al. (1999) which is discussed in the following sections. The PK profiles of efalizumab following single IV doses with observed data and model predicted fit are presented in Figure 11. Mean clearance (CL) decreased from 380 mL/kg/d to 6.6 mL/kg/d for doses of 0.03 mg/ kg to 10 mg/kg, respectively. The volume of distribu-tion of the central compartment (Vc) of efalizumab was 110 mL/kg at 0.03 mg/kg (approximately twice the plasma volume) and decreased to 58 mL/kg at 10mg/kg (approximately equal to plasma volume), consistent with saturable binding of efalizumab to CD11a in the vascular compartment. Because of efalizumab’s non-linear PK, its half-life (t1/2) is dose-dependent.
In a Phase II study of efalizumab, it was shown that at a weekly dosage of 0.1 mg/kg IV, patients did not maintain maximal down modulation of CD11a expression and did not maintain maximal saturation. Also at the end of 8 weeks of efalizumab treatment, 0.1 mg/kg/wk IV, patients did not have statistically significant histological improvement and did not achieve a full clinical response. The minimum weekly IV dosage of efalizumab tested that produced histolo-gical improvements in skin biopsies was 0.3 mg/kg/ wk and this dosage resulted in submaximal saturation of CD11a binding sites but maximal down-modulation of CD11a expression. Improvements in patients’ psoriasis were also observed, as determined by histology and by the Psoriasis area and severity index (PASI) (Papp et al., 2001).
Determination of SC Doses
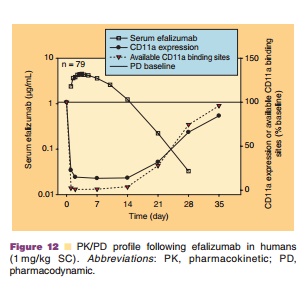
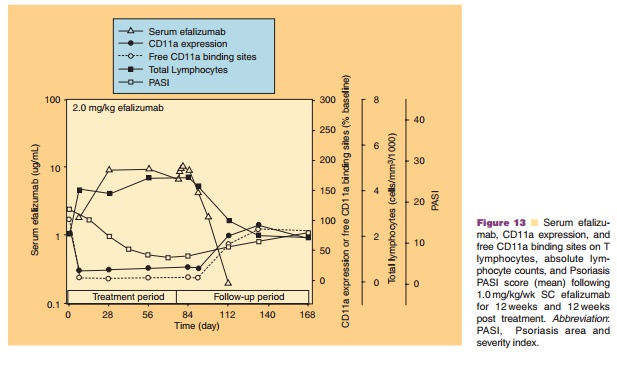
Although efficacy was observed in Phase I and Phase II studies with 0.3 mg/kg/wk IV efalizumab, dosages of 0.6 mg/ kg/wk and greater (given for 7 to 12 weeks) provided more consistent T lymphocyte CD11a saturation and the maximal PD effect. At dosages ≤0.3 mg/kg/wk, large between subject variability was observed, whereas at dosages of 0.6 or 1.0 mg/kg/wk, patients experienced better improvement in PASI scores, with lower between-patient variability in CD11a saturation and down-modulation. Therefore, this dosage was used to estimate an appropriate minimum SC dose of 1 mg/kg/week (based on a 50% bioavailability) that would induce similar changes in PASI, PD measures, and histology. The safety, PK, and PD of a range of SC efalizumab doses (0.5–4.0 mg/kg/ wk administered for 8–12 weeks) were evaluated initially in 2 Phase I studies (Gottlieb et al., 2003). To establish whether a higher SC dosage might produce better results, several Phase III clinical trials assessed a 2.0 mg/kg/wk SC dosage in addition to the 1.0 mg/ kg/wk dosage. A dose of 1.0 mg/kg/wk SC efalizu-mab was selected as it produced sufficient trough levels in patients to maintain the maximal down-modulation of CD11a expression and binding-site saturation between weekly doses (Joshi et al., 2006). Figure 12 depicts the serum efalizumab levels, CD11a expression, and available CD11a binding sites on T-lymphocytes (mean ± SD) after subcutaneous admin-istration of 1 mg/kg efalizumab.
SC Administration of Efalizumab
The PK of SC efalizumab has been well characterized following multiple SC doses of 1.0 and 2.0 mg/kg/wk (Mortensen et al., 2005; Joshi et al., 2006). A Phase I study that collected steady-state PK and PD data for 12 weekly SC doses of 1.0 and 2.0 mg/kg in psoriasis patients, provided most of the pharmacologic data relevant to the marketed product. Although peak serum concentration after the last dose (Cmax) was observed to be higher for the 2.0 mg/kg/wk (30.9 µg/ mL) than for the 1.0 mg/kg/wk dosage (12.4 µg/mL), no additional changes in PD effects were observed at the higher dosages (Mortensen et al., 2005). Following a dose of 1.0 mg/kg/wk, serum efalizumab concen-trations were adequate to induce maximal down-modulation of CD11a expression and a reduction in free CD11a binding sites on T-lymphocytes (Fig. 13). Steady state serum efalizumab levels were reached more quickly with the 1.0 mg/kg/wk dosage at 4 weeks compared with the 2.0 mg/kg/wk dosage at 8 weeks (Mortensen et al., 2005), which is in agreement with the average effective half-life for SC efalizumab 1.0 mg/kg/wk of 5.5 days (Boxenbaum and Battle, 1995). The bioavailability was estimated at approximately 50%. Population PK analyses indicated that body weight was the most significant covariate affecting efalizumab SC clearance, thus supporting body weight-based dosing for efalizumab (Sun et al., 2005).
Related Topics