Chapter: Pharmaceutical Drug Analysis: Pharmaceutical Chemicals: Purity and Management
Various Types of Tests for Quantitative Determinations - Pharmaceutical Chemicals: Management
VARIOUS TYPES OF TESTS FOR QUANTITATIVE DETERMINATIONS
In actual practice, it has been observed that different official compendia describe a number of
detailed types of tests with a view to obtain a constant and regular check that
might be possible to maintain the desired degree of optimum purity both in the
pure pharmaceutical substances and the respective dosage-forms made therefrom.
A number of such tests shall be discussed here briefly
with specific examples wherever possible and necessary :
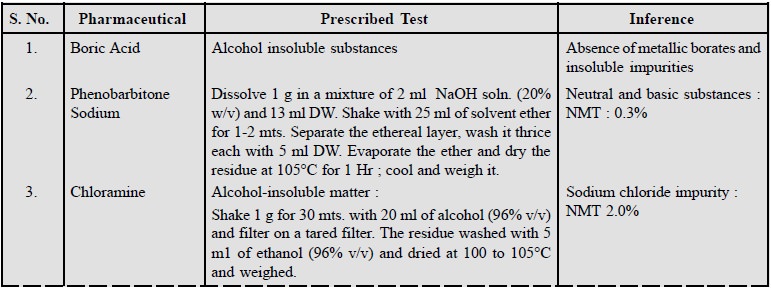
1. Limits of Insoluble Matter
The limits of insoluble matter present in pharmaceutical
substances and stated in various official
com-pendia are given below :

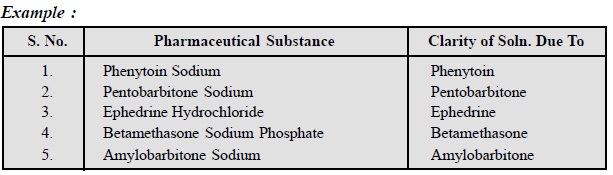
In the same vein, tests for clarity of solution offer
another means of limiting insoluble parent drug sub-stances in their
correspondingly more highly water-soluble derivatives.
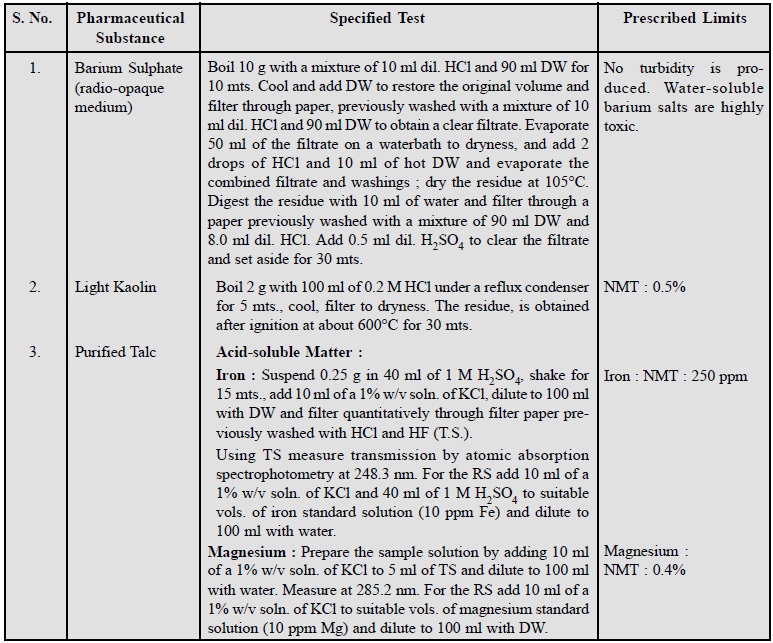
2. Limits of Soluble Matter
In order to detect the presence of some very specific
impurities normally present in the official substances the limits of soluble
impurities have been laid down in different pharmacopoeias. Some typical
examples are cited below :
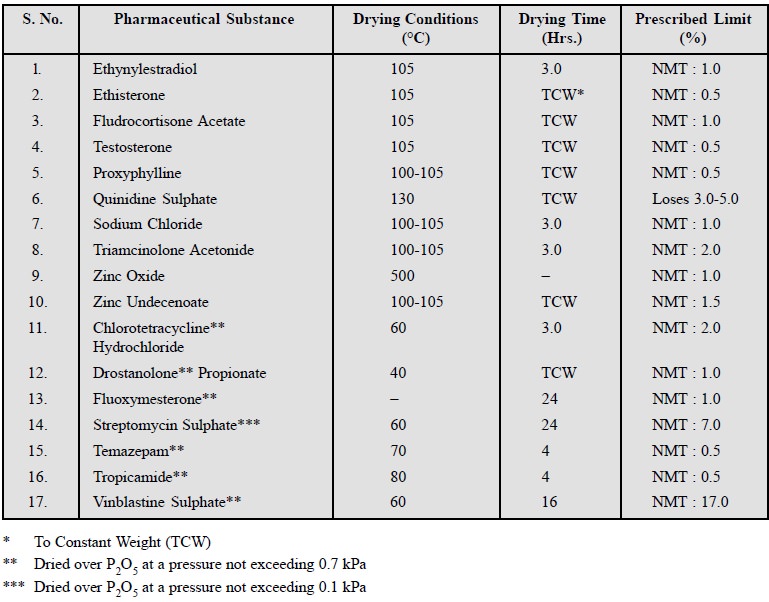
3. Limits of Moisture, Volatile Matter and Residual Solvents
A good number of pharmaceutical substances usually absorb
moisture on storage thereby causing deterioration. Such an anomaly can be
safely restricted and limited by imposing an essential requirement for the loss
in weight (Loss on Drying) when the pharmaceutical chemical is subjected to
drying under specified conditions. The quantum of heat that may be applied to
the substance varies widely as per the following norms :
(a) Nature of
the substance
(b)
Decomposition characterisics of the substance.
Various official
compendia recommended different temperatures and duration of drying either
at atmos-pheric or reduced pressure (vacuum). A few typical examples are stated
below :
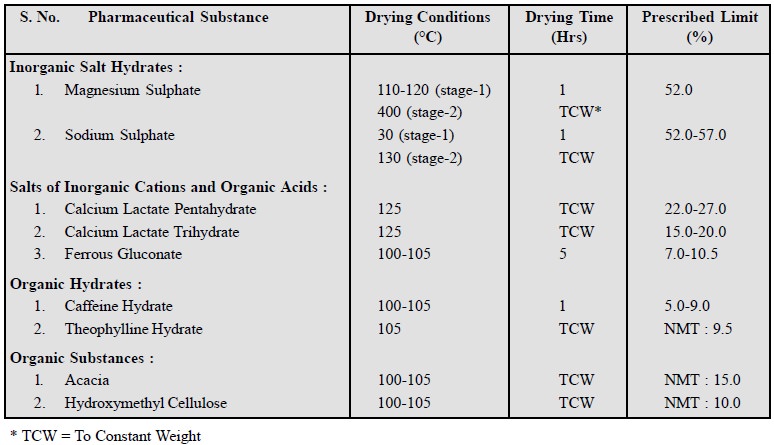
There are four
types of hydrates which may be observed amongst the pharmaceutical chemicals,
namely :
(a) Inorganic
Salt Hydrates e.g., Magnesium
Sulphate (MgSO4.7H2O) ; Sodium Sulphate (Na2SO4.
10 H2O).
(b) Salts of
Inorganic Cations and Organic Acids e.g.,
Calcium Lactate, Ferrous Gluconate.
(c) Organic
Hyrates e.g., Caffeine Hydrate,
Theophylline Hydrate.
(d) Organic
Substances e.g., Acacia,
Hydroxymethyl Cellulose.
These substance either lose all or part of their water of
crystallization on drying which sometimes attains a considerable value as could
be seen in the following data :
3.1. Aquametry
It refers to the determination of water content
titrimetrically with Karl Fischer
Reagent (KFR). This technique has been used exclusively for the
determination of water content in a number of pharmaceutical substances listed
below :
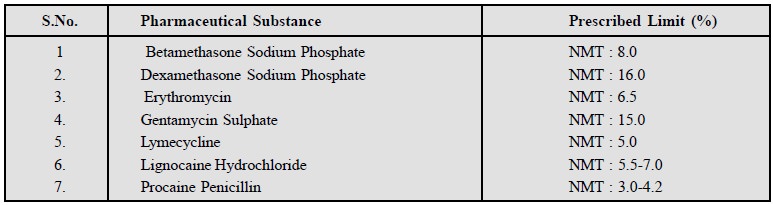
Since the introduction of Gas-Liquid-Chromatography (GLC)
as an essential analytical tool, it has been judiciously exploited as an useful
alternative means for not only determining water content in pharmaceutical
chemicals but also limiting specific volatile substances present in them. It
may be expatiated with the help of the following examples :
Examples :
(i) For Determination of Water Content :
Gonadorelin : (Limit NMT : 7.0 % w/v)
Procedure : Carry out the method for gas
chromatography employing the following solutions
Solution (1) : Dilute 50 ╬╝ l of anhydrous methanol (internal standard) with
sufficient
anhydrous propan-2-ol to produce 100 ml.
Solution (2) : Dissolve 4 mg of the
sample in 1 ml of anhydrous propan-2-ol.
Solution (3) : Dissolve 4 mg of the
sample in 1 ml of solution (1) above.
Solution (4) : Add 10 ╬╝ l of water to 50 ╬╝ l of solution (1).
The chromatographic procedure may be carried out by
employing :
(a) A
stainless-steel column (1 m ├Ś 2 mm) packed with porous polymer beads e.g.,
Chromosorb 102 (60 to 80 mesh) and maintained at 120┬░C.
(b) Helium as the carrier gas.
(c) A Thermal
Conducting Detector (TCD) maintained at 150┬░C. From the chromatograms obtained
and taking into account any water detectable in solution
(1), calculate the percentage w/w of water taking 0.9972
as its weight per ml at 20┬░C.
(ii) For
Limiting Specific Volatile Substance :
Orciprenaline Sulphate : (Limit of Water and Methanol :
6.0% w/w)
Procedure : Perform the method for
gas-chromatography using the following three solutions in water containing :
Solution (1) : 0.50% v/v of MeOH and
0.50% v/v of EtOH (96% v/v)ŌĆöas Internal Standard
Solution (2) : 10% w/v of the sample
Solution (3) : 10% w/v of the sample
and 0.50% v/v of the internal standard.
The chromatographic procedure may be performed using a glass
column (1.5 ├Ś 4 mm) packed with porous polymer beads (80 to 100 mesh) e.g., Porapack-Q and maintained at
140┬░C.
Calculate the percentage w/v of methanol taking 0.792 as
its weight per ml at 20┬░C.
4. Limits of Non-Volatile Matter
Pharmaceutical chemicals belonging to the domain of
inorganic as well as organic substances containing readily volatile matter for
which the various official compendia
prescribe limits of non-volatile matter. It is pertinent to mention here that
the Pharmacopoeia usually makes a clear distinction between substances that are
readily volatile and substances that are volatile upon strong ignition, for
instance :
(a) Readily Volatile : e.g., Organic SubstancesŌĆöalcohol
(95% v/v), isopropyl alcohol, chloroform, halothane, anaesthetic ether,
chlorocresol and trichloroethylene ; and Inorganic
substancesŌĆöammonia solution, hydrogen peroxide solution, water for
injection.
(b) Volatile Upon Strong Ignition : e.g., hydrous wool fat (lanolin).
5. Limits of Residue on Ignition
In fact, the limits of residue on ignition are basically
applicable to the following two categories of pharmaceutical substances, namely
:
(a) Those which
are completely volatile when ignited e.g.,
Hg.
(b) Those which
undergo total decomposition thereby leaving a residue with a definite
composition e.g., calamineŌĆöa basic
zinc carbonate that gives rise to ZnO as the residue.
According to BP, 68.0 to 74.0%
when ignited at a temperature not lower than 900┬░C until, after further
ignition, two successive weighings do not differ, by more than 0.2% of the
weight of the residue.
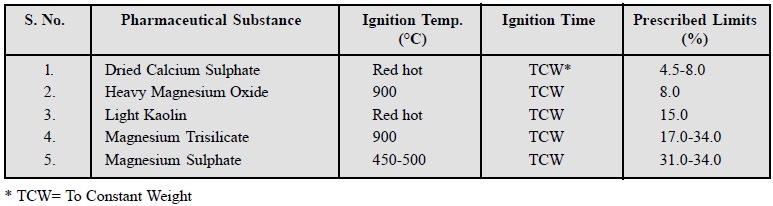
6. Limits of Loss on Ignition
Official compendia include the limits of ŌĆśloss on
ignitionŌĆÖ which is generally applied to relatively stable pharmaceutical substances that are likely to contain thermolabile
impurities. A few typical examples are stated below :
7. Limits on Ash Value
The ash values usually represent the inorganic residue
present in official herbal drugs and pharmaceuti-cal substances. These values
are categorized into four heads,
namely :
(a) Ash Value
(Total Ash),
(b)
Acid-Insoluble Ash,
(c) Sulphated
Ash, and
(d)
Water-Soluble Ash.
These values would be explained with the help of some
typical examples stated below :
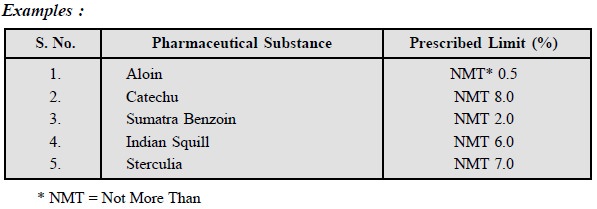
7.1. Ash Value (Total Ash)
Ash value normally designates the presence of inorganic
salts e.g., calcium oxalate found
naturally in the drug, as well as inorganic matter derived from external
sources. The official ash values are of prime importance in examination of the
purity of powdered drugs as enumerated below :
(i) To detect
and check adulteration with exhausted drugs e.g.,
ginger.
(ii) To detect
and check absence of other parts of the plant e.g., cardamom fruit.
(iii) To detect
and check adulteration with material containing either starch or stone cells
that would modify the ash values.
(iv) To ensure
the absence of an abnormal proportion of extraneous mineral matter incorporated
acciden-tally or due to follow up treatment or due to modus operandi at the time of collection e.g., soil, floor sweepings and sand.
The most common procedure recommended for crude drugs is described below :
Procedure : Incinerate 2 to 3 g of the
ground drug in a tared platinum or silica dish at a temperature not exceeding 450┬░C until free from
carbon. Cool and weigh. If a carbon-free ash cannot be obtained in this way,
exhaust the charred mass with hot water (DW), collect the residue on an ashless
filter paper, incinerate the residue and filter paper, add the filtrate,
evaporate to dryness and ignite at a temperature not exceeding 450┬░C. Calculate
the percentage of ash with reference to the air-dried drug.
7.2. Acid-Insoluble Ash
The method described above for ŌĆśtotal ashŌĆÖ present in crude drugs containing calcium oxalate has
certain serious anomalies, namely :
┬Ę
Offers variable results upon ashing based on the
conditions of ignition.
┬Ę
Does not detect soil present in the drug efficaciously.
┬Ę
The limits of excess of soil in the drug are not quite
definite.
Hence, the treatment of the ŌĆśtotal ashŌĆÖ with acid virtually leaves silica exclusively and thus
comparatively forms a better test to detect and limit excess of soil in the
drug than does the ash.
The common procedure usually adopted for the
determination of ŌĆśacid insoluble ashŌĆÖ is given below :
Procedure : Place the ash, as described
earlier, in a crucible, add 15 ml DW and l0 ml hydrochloric acid ( ~ŌĆō 11.5 N), cover with a
watch-glass, boil for 10 minutes and allow to cool. Collect the insoluble
matter on an ashless filtre paper, wash with hot DW until the filtrate is
neutral, dry, ignite to dull redness, allow to cool in a desiccator and weigh.
Repeat until the difference between two successive weighings is not more than l
mg. Calculate the percentage of acid-insoluble ash with reference to the
air-dried drug.
A few typical examples are
listed below :
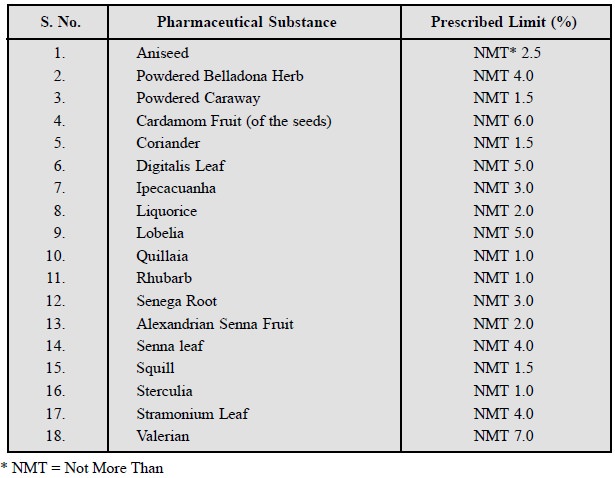
7.3. Sulphated Ash
The estimation of ŌĆśsulphated
ashŌĆÖ is broadly employed in the case of :
(a) Unorganized
drugs e.g., colophony, podophyllum
resin, wool alcohols, wool fat and hydrous wool fat.
(b)
Pharmaceutical substances contained with inorganic impurities e.g.,
Natural Origin : Spray-dried acacia, Frangula
Bark, Activated Charcoal
Organic Substances : Cephalexin, Lignocaine
hydrochloride, Griseofulvin, Diazoxide, Medazapam, Saccharin.
Inorganic Substances : Ammonium chloride, Hydroxy
urea.
The general method for the determination of ŌĆśsulphated ashŌĆÖ is enumerated below :
Procedure : Heat a silica or platimum
crucible to redness for 30 minutes, allow to cool in a desiccator and weigh. Place a suitable quantity
of the substance being examined, accurately weighed in the crucible, add 2 ml
of 1 M sulphuric acid and heat, first on a waterbath and then cautiously over a
flame to about 600┬░C. Continue heating until all black particles have
disappeared and then allow to cool. Add a few drops of 1 M sulphuric acid, heat
to ignition as before and allow to cool. Add a few drops of a 16% solution of
ammonium carbonate, evaporate to dryness and cautiously ignite. Cool, weigh,
ignite for 15 minutes and repeat the procedure to constant weight.
Following are the examples to
depict the ŌĆśsulphated ashŌĆÖ present in
various official pharmaceutical chemicals :
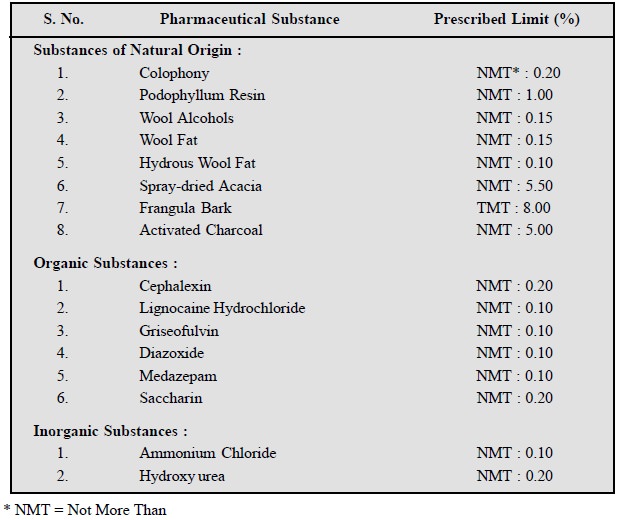
7.4. Water-Soluble Ash
Water-soluble ash is specifically useful in detecting
such samples which have been extracted with water.
A detailed procedure as per the official compendium is
enumerated below :
Procedure : The ash as described earlier,
is boiled for 5 minutes with 25 ml DW, collect the insoluble matter in a sintered-glass crucible or
on an ashless filter paper, wash with hot DW and ignite for 15 minutes at a
temperature not exceeding 450┬░. Subtract the weight of the residue thus
obtained from the weight of the ash.The difference in weight represents the
water-soluble ash. Now, calculate the percentage of water-soluble ash with
reference to the air-dried drug.
A typical example of an official drug is that of
ŌĆśGingerŌĆÖ, the water-soluble ash of which is found to be not more than 6.0%.
Related Topics